Volume 21, Number 2—February 2015
Research
Evidence for Elizabethkingia anophelis Transmission from Mother to Infant, Hong Kong
Cite This Article
Citation for Media
Abstract
Elizabethkingia anophelis, recently discovered from mosquito gut, is an emerging bacterium associated with neonatal meningitis and nosocomial outbreaks. However, its transmission route remains unknown. We use rapid genome sequencing to investigate 3 cases of E. anophelis sepsis involving 2 neonates who had meningitis and 1 neonate’s mother who had chorioamnionitis. Comparative genomics revealed evidence for perinatal vertical transmission from a mother to her neonate; the 2 isolates from these patients, HKU37 and HKU38, shared essentially identical genome sequences. In contrast, the strain from another neonate (HKU36) was genetically divergent, showing only 78.6% genome sequence identity to HKU37 and HKU38, thus excluding a clonal outbreak. Comparison to genomes from mosquito strains revealed potential metabolic adaptations in E. anophelis under different environments. Maternal infection, not mosquitoes, is most likely the source of neonatal E. anophelis infections. Our findings highlight the power of genome sequencing in gaining rapid insights on transmission and pathogenesis of emerging pathogens.
Microbial genome sequencing can enhance diagnosis and control of infectious diseases (1,2). Its ultimate molecular resolution is superior to other phenotypic and genotypic tests and enables not only rapid microbial identification but also characterization of transmission events. The technique has been applied in large-scale infectious disease outbreaks such as those caused by Escherichia coli O104:H4, Staphylococcus aureus, Streptococcus pyogenes, Enterococcus faecium, Pseudomonas aeruginosa, Vibrio cholerae, and mycobacteria (3–14). However, the routine application of this method in diagnostic microbiology and infection control, especially for less well-defined, emerging pathogens, is yet to be explored.
Elizabethkingia anophelis is a recently discovered bacterium isolated from the midgut of the Anopheles gambiae mosquito in 2011 (15). The genus Elizabethkingia also includes E. meningoseptica (previously named Chryseobacterium/Flavobacterium meningosepticum) and E. miricola (16). E. meningoseptica causes neonatal sepsis and infections in immunocompromised persons. E. anophelis has also recently been reported to cause neonatal meningitis in the Central African Republic, and a nosocomial outbreak was reported in an intensive care unit in Singapore (17–19). However, the role of mosquitoes or other sources in the transmission of E. anophelis remains unclear.
In 2012, we encountered 3 cases of Elizabethkingia sepsis associated with meningitis in 2 neonates and chorioamnionitis in a neonate’s mother in a hospital in Hong Kong. Three strains of Elizabethkingia-like, gram-negative bacilli sharing similar phenotypic characteristics were isolated from the 3 patients, but confident identification results were not obtained by matrix-assisted laser desorption ionization/time-of-flight (MALDI-TOF) mass spectrometry and 16S rRNA gene sequencing. Moreover, clinical and microbiological data did not provide adequate clues about the possible transmission route. We therefore attempted to use draft genome sequencing to rapidly dissect transmission pathways and confirm the identity of the species.
Setting and Patients
The 3 patients were hospitalized in an acute regional hospital, Pamela Youde Nethersole Eastern Hospital, which is situated in the eastern area of Hong Kong Island. This study was approved by the Institute Review Board, Hospital Authority, Hong Kong (reference HKEC-2013-051).
Microbiological Methods
Bacterial cultures and phenotypic identification were performed according to standard protocols by using the Vitek II system (bioMérieux, Marcy l’Etoile, France). Antimicrobial drug susceptibility testing was performed by E-test method for vancomycin and Kirby-Bauer disk diffusion for other drugs; because interpretative criteria for Elizabethkingia were lacking, results were interpreted according to Clinical and Laboratory Standards Institute for Pseudomonas aeruginosa (20). MALDI-TOF mass spectrometry was performed by the direct transfer method as described previously (21), with modifications by using the Bruker Daltonics microflex LT system with Reference Library Biotyper version 3.1 (Bruker Daltonik GmbH, Leipzig, Germany). Full 16S rRNA gene amplification and sequencing were performed according to previously published protocols with modifications (22,23). Pulsed-field gel electrophoresis (PFGE) was performed by using the CHEF Mapper XA system (Bio-Rad, Hercules, CA, USA) and restriction endonuclease XbaI as described previously (8,22).
Draft Genome Sequencing and Analysis
The draft genome sequences of the 3 E. anophelis strains were determined by high-throughput sequencing with the Illumina HiSeq 2500 system (Illumina, San Diego, CA, USA). Samples of 50 ng of genomic DNA were extracted by using a genomic DNA purification kit (QIAGEN, Hilden, Germany) from cultures grown overnight on blood agar at 37°C, as described previously (24,25). Each sample was sequenced by 151-bp paired-end reads with mean library size of 350 bp. Sequencing errors were corrected by k-mer frequency spectrum analysis using SOAPec (http://soap.genomics.org.cn/about.html). De novo assembly was performed in SOAPdenovo2 (http://soap.genomics.org.cn/soapdenovo.html). Prediction of protein coding regions and automatic functional annotation was performed by using Glimmer3 (26) and the RAST (Rapid Annotations using Subsystem Technology) server (27). Antibiotic resistomes were identified by using the Antibiotic Resistance Genes Database (28). BLASTn comparisons were run in BLAST+ (http://blast.ncbi.nlm.nih.gov/Blast.cgi) with an E-value cutoff of 10.0. In addition, manual annotation was performed on putative virulence and antibiotic resistance genes by protein domain predictions and multiple sequence alignments with orthologous genes. Intergenomic distance was calculated by using Genome-to-Genome Distance Calculator 2.0 (http://ggdc.dsmz.de/distcalc2.php) (29).
Patients
Figure 1

Figure 1. Clinical course of illness in 3 patients infected with Elizabethkingia anophelis in whom sepsis developed and the mother of patient 1, who had culture-negative postpartum fever, Hong Kong, 2012. Locations where...
In July 2012, a 21-day-old male neonate (patient 1) was admitted to Pamela Youde Nethersole Eastern Hospital for fever of 1 day’s duration. He was born at the same hospital 21 days earlier at 41 weeks’ gestation by vaginal delivery and was discharged on day 3. Physical examination did not show obvious infective focus. Serum C-reactive protein (CRP) was elevated to 109 mg/L. Lumbar puncture was performed; analysis of cerebrospinal fluid (CSF) showed polymorph pleocytosis, elevated protein levels, and low glucose levels (Table). Treatment was initiated for bacterial meningitis with empirical intravenous ampicillin and cefotaxime. Blood and CSF cultures recovered a gram-negative bacillus, designated HKU36. Antimicrobial drugs were changed to vancomycin, piperacillin, and rifampin on day 3. The patient was discharged after 3 weeks of intravenous drug treatment, without neurologic sequelae (Figure 1). The neonate’s mother was admitted to the same hospital 1 day after the infant’s admission for postpartum fever, chills, rigor, and abdominal pain. Transvaginal ultrasound showed no retained gestational products. Serum CRP level was elevated to 109 mg/L; however, blood cultures were negative. She was treated with intravenous cefuroxime and metronidazole and discharged on day 6 with oral cefuroxime and metronidazole.
In November 2012, a 33-year-old woman in week 30 of pregnancy (patient 2) was admitted to the same hospital because of prolonged premature rupture of membranes. She stayed at the same antenatal ward and in the same cubicle as the mother of patient 1 (Figure 1). Fever developed in the patient 3 days after admission, and clinical tests showed peripheral leukocytosis with neutrophilia (Table). Serum CRP was elevated to 108 mg/L. Treatment with intravenous penicillin G was commenced, and an emergency lower segment cesarean section was performed. Placental and uterine swab cultures recovered a gram-negative bacillus, designated HKU37. Blood cultures were negative. Antimicrobial drug treatment was changed to cefuroxime and metronidazole, followed by oral ciprofloxacin for 1 week. Her fever subsided, and she was discharged on day 8.
The baby girl (patient 3) of patient 2 was pale and flaccid at birth; apnea of prematurity developed, requiring cardiopulmonary resuscitation. Peripheral leukopenia and metabolic acidosis were also detected, and serum CRP level was elevated to 70.6 mg/L. Chest radiograph showed bilateral ground-glass appearance. Lumbar puncture was performed, and CSF showed lymphocytic pleocytosis with elevated protein levels and low glucose levels (Table). Ultrasound of the brain showed grade I to II intraventricular hemorrhages. Empirical intravenous ampicillin and cefotaxime at meningitic dose was started. Blood and CSF cultures recovered a gram-negative bacillus, designated HKU38. Antimicrobial drug therapy was changed to intravenous vancomycin, piperacillin/tazobactam, and rifampin on day 3, continuing for 3 weeks. Necrotizing enterocolitis and neonatal jaundice developed, but both resolved with treatment (Figure 1). The infant was discharged on day 54 without neurologic sequelae.
Clinical and Microbiological Investigations
The 3 isolates from these patients, HKU36–38, were nonmotile, oxidase-positive, non–glucose-fermenting, gram-negative bacilli. Their phenotypic characteristics are summarized in the Table and Technical Appendix Table 1. The isolates were identified as E. meningoseptica by using the Vitek II identification system (bioMérieux, Marcy L’Étoile, France). However, MALDI-TOF mass spectrometry identified strains HKU37 and HKU 38 as E. meningoseptica (best match to E. meningoseptica strain 002_NEB14 NFI, with scores of 2.106 and 2.007, respectively), whereas strain HKU36 was only identified to the genus level as Elizabethkingia species (best match to E. meningoseptica strain 002_NEB14 NFI, with score of 1.853) (Technical Appendix Figure 1). The isolates’ 16S rRNA gene sequences exhibited 99.1%–99.9% nucleotide identities to those of E. anophelis type strain R26T (GenBank accession no. EF426425) and 97.4%–99.9% nucleotide identities to those of E. meningoseptica strains deposited in GenBank (GenBank accession nos. HM056770.1, GU180602.1, JQ673498.1, FJ816020, AVCQ01000012, FJ839441.1, JN201943.1, and AJ704540).
The high sequence identities to both E. anophelis and E. meningoseptica made the species identity of the 3 strains uncertain, despite 16S rRNA gene sequencing. Moreover, the strains exhibited minor differences in phenotypes and antibiogram (Table). Further, because the mothers stayed in the same ward before delivery (although 4 months apart), concerns of a possible nosocomial outbreak were raised. However, environmental and water samples from the hospital and patients’ homes were culture-negative for E. anophelis. A program of enhanced infection control measures was enforced in the hospital, and no further cases were identified.
Genome Sequencing and Comparative Analysis of E. anophelis Genomes
Figure 2
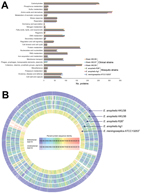
Figure 2. Comparison of draft genome sequence data of the 3 Elizabethkingia anophelis strains from patients in Hong Kong (HKU36–38), E anophelis type strain R26T, and E. meningoseptica type strain ATCC 13253T. A)...
Figure 3

Figure 3. Phylogenetic trees constructed by using draft genome sequences and concatenated sequences of 69 housekeeping genes of 3 Elizabethkingia anophelis strains from patients in Hong Kong (HKU36–38). A) Neighbor-joining tree constructed on...
We sequenced the draft genomes of strains HKU36–38 to investigate their genetic relatedness and confirm their species identity. Sequencing generated 11–15 million paired-end reads per strain (estimated 410–540-fold coverage). After de novo assembly, the 3 draft genomes ranged from 3.92–3.99 Mb in length (G + C content 35.4%–35.8%) and were distributed in 42–52 large (>500 bp) contigs (EMBL accession nos. CBYD010000001–CBYD010000042, CBYE010000001–CBYE010000032, CBYF010000001–CBYF010000038; Technical Appendix Table 2). These contigs contained 3,654–3,667 predicted protein-coding genes (Figure 2, panel A). Using Genome-to-Genome Distance Calculator for intergenomic distance estimation, which enabled genome-based species delineation analogous to traditional DNA–DNA hybridization method, we found that these genomes shared 78.3%–85.4% nucleotide identities to the draft genome sequence of E. anophelis type strain R26T, the initial isolate from an Anopheles gambiae mosquito (GenBank accession no. NZ_ANIW00000000.1). However, the genomes shared only 23.6%–23.7% nucleotide identities to the draft genome sequence of E. meningoseptica type strain ATCC 13253T (GenBank accession no. BARD00000000.1) (Figure 2, panel B). Phylogenetic analysis using the draft genomes and concatenated sequences of 69 housekeeping genes also supported the identification of the 3 strains as E. anophelis (Figure 3; Technical Appendix Figure 2).
Figure 4

Figure 4. Pulsed-field gel electrophoresis (PFGE) analysis of samples from patients in Hong Kong showing 3 Elizabethkingia anophelis strains compared with reference Elizabethkingia isolates. A) PFGE performed by using CHEF Mapper XA system...
The sequences from 52 contigs of strain HKU37 demonstrated 99.4% nucleotide identity to those from 46 contigs of strain HKU38, indicating that these draft genomes are essentially identical (Figure 2, panel B, and Figure 3). The small intergenomic distance can be explained by slight differences in coverage or contig assembly; sequences of 2,000 high-coverage protein-coding genes were identical between HKU37 and HKU38. In contrast, these sequences demonstrated only 78.6% nucleotide identity to those from the 42 contigs of strain HKU36, indicating that strain HKU36 is genetically divergent (Figure 2, panel B, and Figure 3), consistent with PFGE patterns (Figure 4). Moreover, a potential genetic island consisting of conjugative transposable elements was found in strains HKU37 and HKU38 but not in HKU36. Our results exclude a clonal outbreak, but the extremely close genetic relatedness between strains HKU37 and HKU38 provides evidence for vertical transmission from patient 2 to patient 3 (mother to infant).
Potential Virulence Factors and Resistance Genes in E. anophelis
The association of E. anophelis with neonatal meningitis in this and previous reports (17,18) suggests that the bacterium may possess virulence factors that enable it to invade the central nervous system. The 3 draft genomes we identified contain homologs of several virulence genes found in Listeria monocytogenes, which also causes neonatal meningitis. These genes include cell wall hydrolase A, which enables host cell invasion; phosphatidylinositol-specific phospholipase (PlcA) and listeriolysin O (LLO), which enable escape from the primary vacuole of macrophages, and genes that enable survival in the secondary vacuole of macrophages; and virulence cluster protein B (VclB). Phosphatidylinositol-specific phospholipase, listeriolysin O, and virulence cluster protein B are located in the Listeria pathogenicity island LIPI-1 (30,31). Moreover, the 3 genomes we identified contain homologs of arylsulfatase and genes that enable invasion of brain endothelial cells, which contribute to the ability of Escherichia coli to cross the blood–brain barrier in neonatal meningitis (32).
Vertical transmission of E. anophelis from mother to infant also suggests that the bacterium may be able to colonize the vagina before causing ascending chorioamnionitis in the mother and neonatal infection through transplacental spread. A homologof the gene encoding agmatine deiminase, AgDI, which mediates acid tolerance in L. monocytogenes (33), was found in the E. anophelis genomes. Further studies may investigate the possible role of AgDI and potential adherence factors for vaginal colonization in E. anophelis.
Similar to E. meningoseptica, the 3 E. anophelis isolates we identified are resistant to multiple antimicrobial drugs. We found various antimicrobial resistance genes consistent with their resistance phenotypes, including metallo-β-lactamase (blaGOB-1 and blaB14 in strain HKU36 and a novel blaGOB and blaB1 in strains HKU37 and HKU38) and extended-spectrum β-lactamase (blaACME-1 in strains HKU37 and HKU38 and a potential novel blaACME-1 variant in strain HKU36). A comparison of these β-lactamases to their corresponding orthologs in E. meningoseptica genomes revealed only 74%–85% amino acid identities, indicating that E. anophelis and related bacteria are potential reservoirs of novel β-lactamase genes (19,34,35). Other antimicrobial resistance genes found included multidrug-resistance efflux pumps (ATP binding cassette superfamily, major facilitator superfamily, resistance-nodulation-division families, multidrug and toxic-compound extrusion family) that potentially carry resistance to a variety of compounds; chloramphenicol acetyltransferase; aminoglycoside 6-adenyltransferase; and tetracycline resistant gene. Moreover, a putative tetX gene was also identified; this gene encodes a predicted flavin-dependent monooxygenase with tetracycline/tigecycline-degrading activity, although the 3 strains we identified are only resistant to tetracycline but remained susceptible to other related drugs, including tigecycline.
Comparison of Genomes from Human and Mosquito E. anophelis Strains
E. anophelis strains R26T and Ag1 were isolated from mosquitoes (35). Compared with those strains, the genomes of the 3 strains we identified possessed 33 unique hypothetical proteins. Moreover, the genetic island consisting of conjugative transposable elements found in strains HKU37 and HKU38 was also absent in the mosquito strains. In contrast to the mosquito strains, which possessed genes encoding for xylose isomerase (XylA) and xylulose kinase (XylB), these 2 genes were absent in the 3 strains we identified. This finding may reflect different requirements for sugar metabolism in E. anophelis under different environments. Notably, despite the presence of XylA and XylB, E. anophelis mosquito strain R26T did not produce acid from xylose (15). However, this finding does not exclude the strain’s ability to metabolize xylose, as D-xylulose 5-phosphate, the product of XylA and XylB, can be used as a substrate for the pentose-phosphate pathway. XylA and XylB were also absent in the genome of E. meningoseptica type strain ATCC 13253T, which suggests that mosquito strains of E. anophelis may be evolutionarily distinct from clinical strains of E. anophelis and E. meningoseptica. More genome sequence data from other clinical and environmental strains of E. anophelis may shed light on the ecology, biology, and pathogenesis of E. anophelis.
This study demonstrates the power of draft genome sequencing to rapidly dissect transmission pathways for emerging bacterial infections. Our results showed that vertical perinatal transmission had occurred from patient 2, a pregnant woman who had chorioamnionitis, to patient 3, a neonate who had early onset neonatal meningitis. The infective source for patients 2 and 3 was unlikely to have been patient 1 or his mother. However, we speculate that the mother of patient 1 might also have had E. anophelis chorioamnionitis, as evidenced by postpartum fever and abdominal pain, which resulted in late-onset meningitis in her son owing to fastidious bacterial growth. Although strain HKU36 did not belong to the same clone as strains HKU37/38, a polyclonal outbreak of E. anophelis sepsis in the labor ward, in which case an environmental source is likely, could not be excluded.
The discovery of E. anophelis in mosquito gut has raised suspicion that mosquitoes are the source of neonatal meningitis cases in Africa (17). Although Anopheles mosquitoes are not found in Hong Kong, the role of local mosquitoes as reservoirs for E. anophelis remains unknown. Nonetheless, the vertical transmission demonstrated in 1 neonate makes mosquitoes unlikely as vehicles of transmission in our cases.
Our report provides genomic evidence for vertical transmission in neonatal meningitis. Whereas we cannot ascertain how the mother(s) acquired the infection, our results prompt further work to assess the importance of maternal source in neonatal meningitis caused by E. anophelis and other bacterial agents. Maternal colonization with Lancefield group B streptococcus (GBS) during pregnancy is the primary risk factor for early onset neonatal disease. However, direct microbiological evidence for vertical transmission is seldom available, especially for bacterial agents other than GBS. Further genomic studies may help investigate the role of vertical transmission in neonatal meningitis caused by other bacteria. Current indications for intrapartum antimicrobial drugs prophylaxis have been determined on the basis of risk factors for early onset GBS disease; therefore, intravenous penicillin G or ampicillin is often the standard empirical regimen used. However, if further research determines that the mother may also be a source of transmission for other bacterial agents, broader-spectrum antimicrobial drugs may need to be considered as treatment for intrapartum fever or prolonged rupture of membranes.
E. anophelis is likely an underreported bacterium because it can be easily misidentified as E. meningoseptica, which shares a similar phenotypic profile (17,19). The E. anophelis isolates from the recent outbreak reported in Singapore were initially mistakenly identified as E. meningoseptica (19,36). Of the 3 strains we identified, 2 were misidentified as E. meningoseptica with MALDI-TOF mass spectrometry, the state-of-the-art technology, which is replacing conventional phenotypic identification in diagnostic laboratories. The reason for failure of MALDI-TOF mass spectrometry to identify these strains was that reference E. anophelis strains are lacking in existing diagnostic spectrum databases, as is the case with other less commonly encountered organisms (21).
Although 16S rRNA gene sequencing should provide sufficient resolution, some strains indexed as E. meningoseptica, such as strains G3-1-08 and 502, were actually more closely related to E. anophelis than to E. meningoseptica in their 16S rRNA sequences (Figure 3; Technical Appendix Figure 2) (37). These ambiguous, potentially misidentified strains may cause incorrect interpretations in suspected E. anophelis infections. For example, the sequence of strain HKU36 possessed 99.8% nucleotide identity to that of E. meningingoseptica strain G3-1-08 but only 99.1% nucleotide identity to that of E. anophelis strain R26T. Furthermore, phenotypic tests such as acid production from cellobiose and citrate utilization, previously proposed to be useful for identification of E. anophelis (15), are probably unreliable in differentiating among Elizabethkingia species. For example, E. anophelis strain R26T produces acid from cellobiose, but the 3 strains we identified do not; in addition, E. anophelis strains R26T, HKU37, and HKU38, but not strain HKU36, utilize citrate (Technical Appendix Table 1). Strain HKU36 displayed higher MIC of vancomycin than did strains HKU37 and HKU38 and type strains of E. anophelis, E. meningoseptica, and E. miricola, which correlates with previous reports on variable vancomycin susceptibilities in Elizabethkingia (38,39). The species identity of the 3 strains we identified was only resolved by intergenomic comparison. Inclusion of E. anophelis in MALDI-TOF MS databases and rectification of 16S rRNA gene sequences of Elizabethkingia strains deposited in databases will enable accurate diagnosis of more E. anophelis infections.
The draft genome sequences we identified have enabled rapid exploration of novel β-lactamase and other antimicrobial drug resistance genes and possible virulence genes in E. anophelis, highlighting the potential of genome sequencing in identifying novel drug-resistance mechanisms and guiding treatment regimens for emerging, multidrug-resistant bacteria (25,34,40). Because previous cases of E. anophelis neonatal meningitis have been associated with poor outcomes (17,18), further work to elucidate the pathogenesis and antimicrobial drug resistance patterns of this emerging pathogen may help improve clinical management of illness. The findings of potential genes related to neuroinvasion and acid tolerance and the unique genetic characteristics in clinical strains of E. anophelis compared with mosquito strains may also provide insights on the ability of E. anophelis to adapt to different ecologic niches and cause neonatal infection through vertical transmission.
In conclusion, the genome data we obtained for these cases offered superior discriminatory power that supported appropriate infection control measures. The ability to distinguish different bacterial isolates often has critical implications on practical infection-control management, but different strains of the same bacterial species may not be distinguishable by their phenotypes because they reflect a tiny portion of the microbial genome. With better automation and lower costs, draft genome sequencing, which offers a short turnaround time, may replace existing typing methods such as PFGE or multilocus sequence typing for outbreak investigations.
Dr. Lau is a clinical professor in the Department of Microbiology at The University of Hong Kong. Her research focuses on microbial genomics for studying emerging infectious diseases.
Acknowledgments
We thank Cheung-Hing Foo for technical support in bacterial identification and members of the Centre for Genomic Sciences, The University of Hong Kong, for their technical support in genome sequencing.
This work was supported by Health and Medical Research Fund, Food and Health Bureau, The Government of the Hong Kong Special Administrative Region; Strategic Research Theme Fund, Committee for Research and Conference Grant, and University Development Fund, The University of Hong Kong; the Shaw Foundation; donation from Ms. Eunice Lam; and Consultancy Service for Enhancing Laboratory Surveillance of Emerging Infectious Disease for the HKSAR Department of Health.
References
- Loman NJ, Constantinidou C, Chan JZ, Halachev M, Sergeant M, Penn CW, High-throughput bacterial genome sequencing: an embarrassment of choice, a world of opportunity. Nat Rev Microbiol. 2012;10:599–606 . DOIPubMedGoogle Scholar
- Fournier PE, Drancourt M, Raoult D. Bacterial genome sequencing and its use in infectious diseases. Lancet Infect Dis. 2007;7:711–23 . DOIPubMedGoogle Scholar
- Shah MA, Mutreja A, Thomson N, Baker S, Parkhill J, Dougan G, Genomic epidemiology of Vibrio cholerae O1 associated with floods, Pakistan, 2010. Emerg Infect Dis. 2014;20:13–20 . DOIPubMedGoogle Scholar
- Snyder LA, Loman NJ, Faraj LA, Levi K, Weinstock G, Boswell TC, Epidemiological investigation of Pseudomonas aeruginosa isolates from a six-year-long hospital outbreak using high-throughput whole genome sequencing. Euro Surveill. 2013;18:20611 .PubMedGoogle Scholar
- Harris SR, Cartwright EJ, Török ME, Holden MT, Brown NM, Ogilvy-Stuart AL, Whole-genome sequencing for analysis of an outbreak of methicillin-resistant Staphylococcus aureus: a descriptive study. Lancet Infect Dis. 2013;13:130–6 . DOIPubMedGoogle Scholar
- Walker TM, Ip CL, Harrell RH, Evans JT, Kapatai G, Dedicoat MJ, Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect Dis. 2013;13:137–46. DOIPubMedGoogle Scholar
- Bryant JM, Grogono DM, Greaves D, Foweraker J, Roddick I, Inns T, Whole-genome sequencing to identify transmission of Mycobacterium abscessus between patients with cystic fibrosis: a retrospective cohort study. Lancet. 2013;381:1551–60. DOIPubMedGoogle Scholar
- Tse H, Bao JY, Davies MR, Maamary P, Tsoi HW, Tong AH, Molecular characterization of the 2011 Hong Kong scarlet fever outbreak. J Infect Dis. 2012;206:341–51. DOIPubMedGoogle Scholar
- Brown CC, Olsen RJ, Fittipaldi N, Morman ML, Fort PL, Neuwirth R, Spread of virulent group A Streptococcus type emm59 from Montana to Wyoming, USA. Emerg Infect Dis. 2014;20:679–81 . DOIPubMedGoogle Scholar
- Köser CU, Holden MT, Ellington MJ, Cartwright EJ, Brown NM, Ogilvy-Stuart AL, Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. N Engl J Med. 2012;366:2267–75 . DOIPubMedGoogle Scholar
- Loman NJ, Constantinidou C, Christner M, Rohde H, Chan JZ, Quick J, A culture-independent sequence-based metagenomics approach to the investigation of an outbreak of Shiga-toxigenic Escherichia coli O104:H4. JAMA. 2013;309:1502–10. DOIPubMedGoogle Scholar
- Price JR, Golubchik T, Cole K, Wilson DJ, Crook DW, Thwaites GE, Whole-genome sequencing shows that patient-to-patient transmission rarely accounts for acquisition of Staphylococcus aureus in an intensive care unit. Clin Infect Dis. 2014;58:609–18. DOIPubMedGoogle Scholar
- Pérez-Lago L, Comas I, Navarro Y, González-Candelas F, Herranz M, Bouza E, Whole genome sequencing analysis of intrapatient microevolution in Mycobacterium tuberculosis: potential impact on the inference of tuberculosis transmission. J Infect Dis. 2014;209:98–108 . DOIPubMedGoogle Scholar
- Johnson PD, Ballard SA, Grabsch EA, Stinear TP, Seemann T, Young HL, A sustained hospital outbreak of vancomycin-resistant Enterococcus faecium bacteremia due to emergence of vanB E. faecium sequence type 203. J Infect Dis. 2010;202:1278–86 . DOIPubMedGoogle Scholar
- Kämpfer P, Matthews H, Glaeser SP, Martin K, Lodders N, Faye I. Elizabethkingia anophelis sp. nov., isolated from the midgut of the mosquito Anopheles gambiae. Int J Syst Evol Microbiol. 2011;61:2670–5. DOIPubMedGoogle Scholar
- Kim KK, Kim MK, Lim JH, Park HY, Lee ST. Transfer of Chryseobacterium meningosepticum and Chryseobacterium miricola to Elizabethkingia gen. nov. as Elizabethkingia meningoseptica comb. nov. and Elizabethkingia miricola comb. nov. Int J Syst Evol Microbiol. 2005;55:1287–93. DOIPubMedGoogle Scholar
- Frank T, Gody JC, Nguyen LB, Berthet N, Le Fleche-Mateos A, Bata P, First case of Elizabethkingia anophelis meningitis in the Central African Republic. Lancet. 2013;381:1876. DOIPubMedGoogle Scholar
- Bobossi-Serengbe G, Gody JC, Beyam NE, Bercion R. First documented case of Chryseobacterium meningosepticum meningitis in Central African Republic. Med Trop (Mars). 2006;66:182–4 .PubMedGoogle Scholar
- Teo J, Tan SY, Tay M, Ding Y, Kjelleberg S, Givskov M, First case of E anophelis outbreak in an intensive-care unit. Lancet. 2013;382:855–6. DOIPubMedGoogle Scholar
- Clinical and Laboratory Standards Institute. Performance standards for antimicrobial disk susceptibility tests. Approved standard, 11th ed. M02–A11. Wayne (PA): The Institute; 2012.
- Lau SK, Tang BS, Curreem SO, Chan TM, Martelli P, Tse CW, Matrix-assisted laser desorption ionization–time of flight mass spectrometry for rapid identification of Burkholderia pseudomallei: importance of expanding databases with pathogens endemic to different localities. J Clin Microbiol. 2012;50:3142–3. DOIPubMedGoogle Scholar
- Woo PC, Lau SK, Teng JL, Que TL, Yung RW, Luk WK, L Hongkongensis study group. Association of Laribacter hongkongensis in community-acquired gastroenteritis with travel and eating fish: a multicentre case-control study. Lancet. 2004;363:1941–7. DOIPubMedGoogle Scholar
- Lau SK, Curreem SO, Lin CC, Fung AM, Yuen KY, Woo PC. Streptococcus hongkongensis sp. nov., isolated from a patient with an infected puncture wound and from a marine flatfish. Int J Syst Evol Microbiol. 2013;63:2570–6. DOIPubMedGoogle Scholar
- Tse H, Tsoi HW, Leung SP, Lau SK, Woo PC, Yuen KY. Complete genome sequence of Staphylococcus lugdunensis strain HKU09–01. J Bacteriol. 2010;192:1471–2. DOIPubMedGoogle Scholar
- Woo PC, Lau SK, Tse H, Teng JL, Curreem SO, Tsang AK, The complete genome and proteome of Laribacter hongkongensis reveal potential mechanisms for adaptations to different temperatures and habitats. PLoS Genet. 2009;5:e1000416. DOIPubMedGoogle Scholar
- Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;23:673–9. DOIPubMedGoogle Scholar
- Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. DOIPubMedGoogle Scholar
- Liu B, Pop M. ARDB—antibiotic resistance genes database. Nucleic Acids Res. 2009;37:D443–7 . DOIPubMedGoogle Scholar
- Auch AF, Klenk HP, Göker M. Standard operating procedure for calculating genome-to-genome distances based on high-scoring segment pairs. Stand Genomic Sci. 2010;2:142–8. DOIPubMedGoogle Scholar
- Dussurget O. New insights into determinants of Listeria monocytogenes virulence. Int Rev Cell Mol Biol. 2008;270:1–38.
- Kreft J, Vázquez-Boland JA, Altrock S, Dominguez-Bernal G, Goebel W. Pathogenicity islands and other virulence elements in Listeria. Curr Top Microbiol Immunol. 2002;264:109–25 .PubMedGoogle Scholar
- Hoffman JA, Badger JL, Zhang Y, Huang SH, Kim KS. Escherichia coli K1 aslA contributes to invasion of brain microvascular endothelial cells in vitro and in vivo. Infect Immun. 2000;68:5062–7. DOIPubMedGoogle Scholar
- Cheng C, Chen J, Fang C, Xia Y, Shan Y, Liu Y, Listeria monocytogenes aguA1, but not aguA2, encodes a functional agmatine deiminase: biochemical characterization of its catalytic properties and roles in acid tolerance. J Biol Chem. 2013;288:26606–15. DOIPubMedGoogle Scholar
- Matyi SA, Hoyt PR, Hosoyama A, Yamazoe A, Fujita N, Gustafson JE. Draft genome sequences of Elizabethkingia meningoseptica. Genome Announc. 2013;1:e00444–13.
- Kukutla P, Lindberg BG, Pei D, Rayl M, Yu W, Steritz M, Draft genome sequences of Elizabethkingia anophelis strains R26T and Ag1 from the midgut of the malaria mosquito Anopheles gambiae. Genome Announc. 2013;1:e01030–13.
- Balm MN, Salmon S, Jureen R, Teo C, Mahdi R, Seetoh T, Bad design, bad practices, bad bugs: frustrations in controlling an outbreak of Elizabethkingia meningoseptica in intensive care units. J Hosp Infect. 2013;85:134–40. DOIPubMedGoogle Scholar
- Quick J, Constantinidou C, Pallen MJ, Oppenheim B, Loman NJ. Draft genome sequence of Elizabethkingia meningoseptica isolated from a traumatic wound. Genome Announc. 2014;2:e00355–14.
- Sarma S, Kumar N, Jha A, Baveja U, Sharma S. Elizabethkingia meningosepticum: an emerging cause of septicemia in critically ill patients. J Lab Physicians. 2011;3:62–3. DOIPubMedGoogle Scholar
- Teo J, Tan SY, Liu Y, Tay M, Ding Y, Li Y, Comparative genomic analysis of malaria mosquito vector-associated novel pathogen Elizabethkingia anophelis. Genome Biol Evol. 2014;6:1158–65. DOIPubMedGoogle Scholar
- Holden MT, Feil EJ, Lindsay JA, Peacock SJ, Day NP, Enright MC, Complete genomes of two clinical Staphylococcus aureus strains: evidence for the rapid evolution of virulence and drug resistance. Proc Natl Acad Sci U S A. 2004;101:9786–91. DOIPubMedGoogle Scholar
Figures
Table
Cite This Article1These authors contributed equally to this article.
Table of Contents – Volume 21, Number 2—February 2015
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Patrick C.Y. Woo, Department of Microbiology, The University of Hong Kong, Room 423, University Pathology Building, Queen Mary Hospital Compound, Pokfulam Road, Hong Kong, China
Top