Outbreak of Henipavirus Infection, Philippines, 2014
Paola Katrina G. Ching, Vikki Carr de los Reyes, Maria Nemia Sucaldito, Enrique Tayag, Alah Baby Columna-Vingno, Fedelino F. Malbas, Gilbert C. Bolo, James Sejvar, Debbie Eagles, Geoffrey Playford, Erica Dueger, Yoshihiro Kaku, Shigeru Morikawa, Makoto Kuroda, Glenn A. Marsh, Sam McCullough, and A. Ruth Foxwell

Author affiliations: Department of Health, Manila, Philippines (P.K.G. Ching, V.C. de los Reyes, M.N. Sucaldito, E. Tayag, F.F. Malbas Jr.); Center for Health Development Region XII, General Santos, Philippines (A.B. Columna-Vingno); Department of Agriculture, Manila (G.C. Bolo, Jr); World Health Organization, Manila (J.J. Sejvar, D. Eagles, G. Playford, E. Dueger, A.R. Foxwell); Centers for Disease Control and Prevention, Atlanta, Georgia, USA (J.J. Sejvar, E. Dueger); Australian Animal Health Laboratory, Geelong, Victoria, Australia (D. Eagles, G.A. Marsh, S. McCullough); University of Queensland, Brisbane, Queensland, Australia (G. Playford); National Institute of Infectious Disease, Tokyo, Japan (Y. Kaku, S. Morikawa, M. Kuroda); Australian National University, Canberra, Australian Capital Territory, Australia (A.R. Foxwell)
Main Article
Figure 2
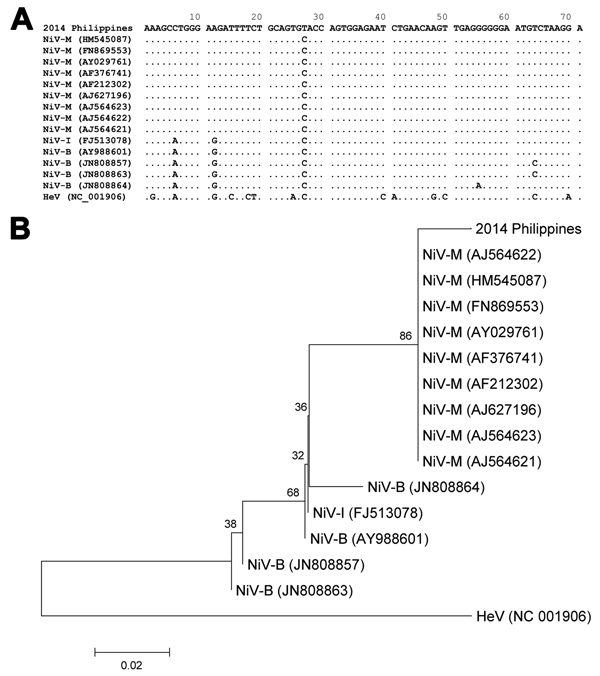
Figure 2. Alignment (A) and phylogenetic relationship (B) of partial phosphoprotein gene sequences (71 mer) of henipaviruses, including the fragment obtained by next-generation sequencing from a patient in Philippines (2014 Philippines). The alignment was conducted by using the MUSCLE program (http://www.ebi.ac.uk/Tools/msa/muscle/), and the phylogenetic tree from these data was constructed by using the neighbor-joining method. The optimal tree with sum of branch length equal to 0.23440320 is shown. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) are shown. The phylogenetic tree is drawn to scale; branch lengths in the same units as those of the evolutionary distances are used to infer the phylogenetic tree. The scale bar represents 0.02 substitutions per site. The evolutionary distances were computed by using the Kimura 2-parameter method and are presented as number of base substitutions per site. The analysis involved 16-nt sequences. All positions containing gaps and missing data were eliminated. The final dataset contained 71 positions. Evolutionary analyses were conducted by using MEGA6 (http://www.megasoftware.net). The accession numbers of each sequence are shown for the viruses. HeV, Hendra virus. NiV-B, Nipah virus Bangladesh strain; NiV-I, Nipah virus Indian strain; NiV-M, Nipah virus Malaysian strain.
Main Article
Page created: January 21, 2015
Page updated: January 21, 2015
Page reviewed: January 21, 2015
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.