Volume 21, Number 5—May 2015
Dispatch
Genetic Characterization of Highly Pathogenic Avian Influenza (H5N8) Virus from Domestic Ducks, England, November 2014
Cite This Article
Citation for Media
Abstract
Genetic sequences of a highly pathogenic avian influenza (H5N8) virus in England have high homology to those detected in mainland Europe and Asia during 2014. Genetic characterization suggests this virus is an avian-adapted virus without specific affinity for zoonoses. Spatio-temporal detections of H5N8 imply a role for wild birds in virus spread.
Aquatic birds are considered to be the natural reservoir of low pathogenicity avian influenza viruses of subtypes H1–H16; in these birds, including ducks, they generally do not cause clinical signs (1). In contrast, highly pathogenic avian influenza (HPAI) viruses of certain H5 and H7 strains cause high death rates in poultry with substantial economic losses and are thought to be derived from low pathogenicity avian influenza viruses of wild bird origin. China, Japan, and South Korea have reported outbreaks of highly related HPAI (H5N8) virus in poultry and migratory birds since early 2014 (2,3). Germany and the Netherlands have reported outbreaks among poultry with closely related H5N8 viruses in turkey, chicken, and duck farms since early November 2014. On November 16, 2014, an outbreak of HPAI (H5N8) virus was confirmed on a duck breeding farm in East Yorkshire, England, UK (4). The premises contained 6,000 breeding ducks ≈60 weeks of age housed in 3 sheds; the ducks showed only mild clinical signs of illness.
Thirty-seven of 120 individual swab samples and 5 of 12 tissue pools (Technical Appendix) were confirmed positive for avian influenza virus by identification of the matrix (M) gene and H5 real-time reverse transcription PCR (rRT-PCR) as described (5). A highly pathogenic pathotype was confirmed by RT-PCR, then Sanger sequencing of the hemagglutinin (HA) gene as described (5). This process showed the polybasic cleavage site PLRERRRKR/GLF from multiple bird swab and tissue samples from each shed. HPAI virus was isolated in 9- to10-day-old specific pathogen–free embryonated hens’ eggs after incubating 2 days as described (6). A primary isolate, A/duck/England/36254/2014 (dkEng14), was obtained after egg inoculation with pooled intestinal samples from 1 shed and was typed as H5N8 by using standard protocols (6). We obtained full genome sequence from dkEng14 by next-generation sequencing (Technical Appendix). Full genome sequences of 2 more isolates, A/duck/England/36038/2014 and A/duck/England/36226/2014, derived from pooled cloacal swabs and pooled viscera from 2 different sheds, respectively, were also assessed by next generation sequencing and analyzed. We entered genome sequences on the Global Initiative on Sharing All Influenza Data EpiFlu database (7) under accession nos. EPI547670– EPI547677, EPI550848–EPI550849, and EPI558000–EPI558013. Initial maximum likelihood phylogenetic analysis of the HA gene was conducted as described (5) and the evolutionary analyses performed by using MEGA6 (8).
Figure 1

Figure 1. Maximum clade credibility tree of 31 H5 sequences derived from the hemagglutinin gene of avian influenza viruses (1,608 nt). Sampling dates and locations are included on the tip labels; where specific...
Phylogenetic analysis showed 99.9%–100% similarity among the 3 duck isolates from the single premises in England and 99.4–100% similarity among HPAI (H5N8) sequences from China, Japan, South Korea, Germany, and the Netherlands; all belonged to Asian lineage H5 clade 2.3.4.4. Sequence comparisons based on 1,608 nt of the HA gene from each of the 3 duck isolates identified A/turkey/Germany/MV-R2472/2014 (H5N8) as the closest match, at 99.8% similarity. We implemented further phylogenetic analysis using Bayesian Markov chain Monte Carlo simulation in the BEAST package version 1.7 (9). The maximum clade credibility tree (Figure 1) had a similar topology to that observed for the maximum-likelihood tree.
The time elapsed between the detection of the isolates in England and the most recent common ancestor (MRCA) of the H5N8 virus cluster from Europe and Japan was ≈5 months (95% highest posterior density [HPD] range 2.7–7.7 months; approximately June 2014). This cluster shares homology of the HA gene with viruses detected in South Korea in early 2014. The ancestor of the viruses from continental Europe, Japan, and Korea occurred ≈13 months (11–15.5 months, 95% HPD; approximately October 2013) before the detection of the English isolates. The separation between the H5N8 HA sequences from England is attributed to 2 nonsynonymous mutations coding for amino acid substitution S181P and H273Y (HA numbering based on the mature H5 protein). Position 181 is in close proximity to the receptor binding site and antigenic site Sb (10); however, the effect of this condition on antigenicity has not yet been defined.
Figure 2
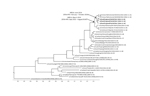
Figure 2. Maximum clade credibility tree of 31 N8 sequences derived from the neuraminidase gene of avian influenza viruses (1377 nt). Sampling dates and locations are included on the tip labels; where specific...
The HA sequence variation observed within 1 premises in which ducks were infected in England is potentially the consequence of virus adaptation within the flock subsequent to a single introduction. Full-genome comparison of the 3 viruses in England showed amino acid differences in the HA and NA genes only (Table 1). Further analyses with sequences from the NA gene indicated a similar most recent common ancestor (≈5 months) for the European lineage. However, each of the 3 virus sequences detected in England grouped together with those from the Netherlands (Figure 2).
To investigate the potential zoonotic affinity of dkEng14, sequence data was compared with the H5N1 genetic changes inventory at the US Centers for Disease Control and Prevention (11) to identify single or collective mutations that might influence viral phenotypic characteristics of importance and may indicate adaptation to mammalian species or alter susceptibility to antiviral drugs. A total of 8 point mutations among 114 of interest were identified (Table 2). In addition to the polybasic (PB) cleavage site in the HA protein, positions 133 and 156 each contained an alanine residue (H5 numbering relative to A/Vietnam/1203/2004), which is reportedly related to increased virus binding to α2,6 sialic acid human receptors.
An asparagine that has been associated with reduced susceptibility to amantadine and rimantadine antiviral drugs was present at position 31 in the M2 protein, but we did not detect any signature motifs associated with resistance to antivirals targeting neuraminidase. Aspartic acid at position 30 and alanine at position 215 in the M1 protein together with serine at position 42 and methionine at position 101 in the nonstructural 1 protein were observed; all have been individually linked to increased virulence in mice. A serine residue at position 66 in the PB1-F2 protein was also observed. This mutation has also been linked to increased virulence in mice and studies in ducks showed a minor role in pathogenesis. Of interest is the lack of a deletion in the nonstructural 1 protein at amino acid positions 80–84 that is conserved among contemporary H5N1 viruses, possibly decreasing the zoonotic potential of the H5N8 virus. Two mutations in PB2, E627K, and K526R (12), described to be involved with mammalian host adaptation and increased replication in mammalian cells, were not observed. Eight mutations were identified in dkEng14; only 1 (T156A) of the 5 necessary mutations associated with high risk for zoonotic transmission were observed (13,14). The mutations identified in Table 2 are analogous to that of A/mallard duck/Korea/W452/2014 (H5N8), a virus that has been studied for its pathogenic and pandemic potential (15) and was found to result in impaired replication and inefficient contact transmission among ferrets.
The genome of the H5N8 virus isolated in England suggests that it is still predominantly an avian-adapted virus, without any specific increased affinity for humans. Close genetic homology among the viral genes of the H5N8 viruses detected in England, the Netherlands, and Germany suggest they share a common ancestor with the recent H5N8 viruses isolated from wild ducks in Japan, a result of reassortment estimated to have occurred in June 2014. Reliable interpretation of the topology of the European and Japanese cluster cannot be made with these similar sequences. Phylogenetic analysis of sequences from more viruses will help to resolve these relationships. Detection of H5N8 (HPAI) viruses in 3 countries in Europe over a short time period in different poultry species without the establishment of clear epidemiologic links implicates a role for wild birds in spreading of viruses. The potential for further dissemination of HPAI (H5N8) viruses in Europe is a threat to poultry. Viral sequence analysis from new outbreaks is recommended to monitor virus evolution, understand risk pathways for introduction, and assess the emergence of mutations that may be relevant for veterinary and public health.
Miss Hanna is a research scientist at the Animal and Plant Health Agency, Weybridge, UK. Her primary research interests are genetic characterization and pathogenesis studies of avian influenza viruses.
Acknowledgments
We thank the avian influenza team at the Animal and Plant Health Agency (APHA-Weybridge). We thank Vanessa Ceeraz, James Seekings, Alejandro Nuñez, Scott Reid, and Vivien Coward for investigative testing, Brandon Löndt for assistance with sequence mutation analysis, and Charlotte Cook for assisting in implementing the BEAST program. We also thank Dan Horton for his guidance with the BEAST analysis.
This work was supported financially by Defra (Project SV3400) and the European Commission through their support of the Avian Influenza EU reference laboratory at APHA.
References
- Hulse-Post DJ, Sturm-Ramirez KM, Humberd J, Seiler P, Govorkova EA, Krauss S, Role of domestic ducks in the propagation and biological evolution of highly pathogenic H5N1 influenza viruses in Asia. Proc Natl Acad Sci U S A. 2005;102:10682–7. DOIPubMedGoogle Scholar
- Fan S, Zhou L, Wu D, Gao X, Pei E, Wang T, A novel highly pathogenic H5N8 avian influenza virus isolated from a wild duck in China. Influenza Other Respir Viruses. 2014;8:646–53.
- Ku KB, Park EH, Yum J, Kim JA, Oh SK, Seo SH. Highly pathogenic avian influenza A(H5N8) virus from waterfowl, South Korea, 2014. Emerg Infect Dis. 2014;20:1587–8. DOIPubMedGoogle Scholar
- World Organisation for Animal Health (OIE). Highly pathogenic avian influenza. Immediate notification report. Report reference: REF OIE 16542, report date: 17/11/2014 [cited 2014 Dec 01]. http://www.oie.int/wahis_2/public/wahid.php/Reviewreport/Review?reportid=16542
- Slomka MJ, To TL, Tong HH, Coward VJ, Hanna A, Shell W, Challenges for accurate and prompt molecular diagnosis of clades of highly pathogenic avian influenza H5N1 viruses emerging in Vietnam. Avian Pathol. 2012;41:177–93. DOIPubMedGoogle Scholar
- European Union. Diagnostic manual for avian influenza. Official Journal of the European Union. 2006 [cited 2014 Dec 01]. http://faolex.fao.org/docs/pdf/eur65757.pdf
- Global Initiative on Sharing Avian Influenza Data [cited 2014 Dec 1]. http://platform.gisaid.org/epi3/frontend#5412a6
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30:2725–9. DOIPubMedGoogle Scholar
- Lu L, Lycett SJ, Leigh Brown AJ. Determining the phylogenetic and phylogeographic origin of highly pathogenic avian influenza (H7N3) in Mexico. PLoS ONE. 2014;9:e107330. DOIPubMedGoogle Scholar
- Siddique N, Naeem K, Abbas MA, Ahmed Z, Malik SA. Sequence and phylogenetic analysis of highly pathogenic avian influenza H5N1 viruses isolated during 2006–2008 outbreaks in Pakistan reveals genetic diversity. Virol J. 2012;9:300. DOIPubMedGoogle Scholar
- Centers for Disease Control and Prevention. Influenza H5N1 genetic changes inventory: a tool for influenza surveillance and preparedness. 2012 [cited 2014 Dec 01]. http://www.cdc.gov/flu/pdf/avianflu/h5n1-inventory.pdf
- Song W, Wang P, Mok BW, Lau SY, Huang X, Wu WL, The K526R substitution in viral protein PB2 enhances the effects of E627K on influenza virus replication. Nat Comm. 2014;5:5509. DOIGoogle Scholar
- Herfst S, Schrauwen EJ, Linster M, Chutinimitkul S, de Wit E, Munster VJ, Airborne transmission of influenza A/H5N1 virus between ferrets. Science. 2012;336:1534–41. DOIPubMedGoogle Scholar
- Imai M, Watanabe T, Hatta M, Das SC, Ozawa M, Shinya K, Experimental adaptation of an influenza H5 HA confers respiratory droplet transmission to a reassortant H5 HA/H1N1 virus in ferrets. Nature. 2012;486:420–8 .DOIPubMedGoogle Scholar
- Kim Y-I, Pascua PNQ, Kwon H-I, Lim G-J, Kim E-H, Yoon S-W, Pathobiological features of a novel, highly pathogenic avian influenza A(H5N8) virus. Emerg Microbes Infect. 2014;3:e75.
Figures
Tables
Cite This ArticleTable of Contents – Volume 21, Number 5—May 2015
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Amanda Hanna, Virology, Animal and Plant Health Agency (APHA-Weybridge), Woodham Lane, Addlestone, Surrey, KT15 3NB, UK
Top