Volume 22, Number 4—April 2016
Dispatch
Neisseria meningitidis Serogroup X in Sub-Saharan Africa
Cite This Article
Citation for Media
Abstract
The epidemiology of meningococcal disease varies by geography and time. Whole-genome sequencing of Neisseria meningitidis serogroup X isolates from sub-Saharan Africa and Europe showed that serogroup X emergence in sub-Saharan Africa resulted from expansion of particular variants within clonal complex 181. Virulence of these isolates in experimental mouse models was high.
The epidemiology of meningococcal disease, caused by infection with the bacterium Neisseria meningitidis, varies substantially by geography and time. The disease can occur as sporadic cases, outbreaks, and large epidemics. Most cases occur in what has been termed the “meningitis belt” in sub-Saharan Africa, where the World Health Organization estimated that 12,464 suspected cases occurred during the 2013 meningitis season and that 1,131 were fatal (case-fatality ratio 9%) (1).
The number of cases was lower during the 2013 season than during previous seasons and can be explained by the introduction of mass vaccination with the meningococcal A conjugate vaccine MenAfriVac (Serum Institute of India, Pune, India) (1). Cyclic epidemics spanned the meningitis belt and were caused mainly by isolates of N. meningitidis serogroup A that belonged to several genetic lineages called clonal complexes (CCs). Molecular typing of meningococci has used multilocus sequence typing (MLST) to determine CCs on the basis of the polymorphism of 7 housekeeping genes (2). Since the 1990s, isolates of other N. meningitidis serogroups, such as serogroup X, have been reported in several countries within the meningitis belt (3). In 2006, high incidence of serogroup X was first reported in Niamey, Niger (4), and thereafter in other countries, reinforcing the need for an effective vaccine against serogroup X (5). Serogroup X has rarely been detected in Europe (6).
MLST may not be able to resolve the variations associated with the emergence of meningococcal isolates. Whole-genome sequencing can provide data that enable allelic comparisons between meningococcal isolates (7,8) and tracing of the emergence and spread of meningococcal isolates.
As part of our mission as a World Health Organization collaborating center, we performed MLST on N. meningitidis serogroup X isolates sent to the Institut Pasteur during 1998–2005 (Table 1). We further performed whole-genome sequencing on 8 invasive serogroup X isolates obtained during 1998–2005 from several countries within the meningitis belt and 3 invasive isolates from France (Table 1).
Genomic DNA was extracted by using a Genomic-tip 20/G kit (QIAGEN, Valencia CA, USA) from culture grown overnight on gonococcal medium base agar plates with Kellogg supplements (9). Whole-genome sequencing was performed by using an Illumina HiSeq 2000 sequencer (Illumina, San Diego, CA, USA) and assembled as described (10). Sequences are available through the PubMLST database (http://pubmlst.org/neisseria/), which runs on the Bacterial Isolate Genome Sequence Database (BIGSdb) platform (11); identification numbers are 34731–34732, 34734–34741, and 34745. We analyzed the 11 isolates from sub-Saharan Africa and France and all other serogroup X genomes available in BIGSdb by using the BIGSdb genome comparator tool.
Figure 1
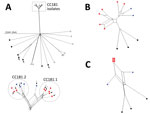
Figure 1. Neighbor-Net SplitsTree graphs generated using SplitsTree4 version 4.13.1 (http://www.splitstree.org) to visualize trees of Neisseria meningitidis serogroup X isolates. A) All 30 Neisseria meningitidis serogroup X isolates available on BIGSdb (<>
The isolates from Africa were genetically related, belonged to CC181, and were separated from all the other isolates that belonged to other CCs (Table 1; Figure 1, panel A). The CC181 isolates formed a single main lineage comprising 2 sublineages, 181.1 and 182.2. Sublineage 181.1 was detected only in isolates obtained in 2006, but sublineage 181.2 was detected in isolates from both periods (1990s and since 2006). Genomes from the 3 isolates from France and those available in the PubMLST database were highly diverse with no close clustering (Figure 1, panel A).
Because we assembled the sequences by using the same method, we further focused on the 11 genomes according to particular groups of genes involved in meningococcal virulence (biosynthesis of the capsule, serogroup B vaccine antigens, lipooligosaccharide, pilin, and iron acquisition). Among genes involved in iron acquisition, the isolates from Africa, unlike those from France, lacked the hpuA and hpuB genes that mediate heme-iron acquisition from hemoglobin and hemoglobin–haptoglobin complexes. However, the isolates from Africa harbored the hemoglobin receptor that was missing in the isolates from France. The hemoglobin receptor is detected more frequently among isolates involved with disease than among those involved with carriage (12). The analysis of the polymorphism of these iron-acquisition genes showed that the isolates from Africa clustered together, separate from the isolates from France (Figure 1, panel B).
Figure 2
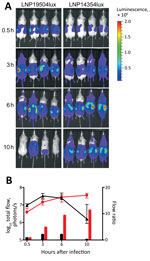
Figure 2. Dynamic imaging showing the multiplication and spread of Neisseria meningitidis in BALB/c transgenic mice expressing the human transferrin. A) Dorsal views of 8 mice (4/group) analyzed for bioluminescence at intervals after...
We tested the virulence of these isolates in a relevant animal model, transgenic mice expressing the human transferrin (13). The animal experiments were conducted in accordance with the European Union Directive 2010/63/EU (and its revision 86/609/EEC) for the protection of animals used for scientific purposes and were approved by the Institut Pasteur Review Board, which is part of the Regional Committee of Ethics of Animal Experiments of the Paris region (permit 99–174). We constructed bioluminescent variants of isolates LNP19504 and LNP14354 by transformation with the previously published recombinant plasmid pDG34 harboring the luxCDABE operon expressed by the PporB meningococcal promoter (14). The structure was recombined in N. meningitidis downstream of the recombinogenic pilE gene to obtain the strains LNP19504lux and LNP14354lux. The insertion of the lux operon did not modify bacterial invasion in this animal model (13). These 2 bioluminescent strains were then used to infect transgenic mice by intraperitoneal injection of 5 × 106 CFU in 500 μL of the corresponding bacterial suspension. Dynamic bioluminescence imaging was used to follow the infection. After 30 minutes of intraperitoneal challenge, bacteria were found mainly in the peritoneal cavity and the image signals did not differ substantially. Bacterial infection then started to spread, enabling detection of bioluminescent signals at distal anatomic sites; signals in the skull and all over the infected mice suggested systemic infection and septicemia (13). However, after 6 hours of infection, the signal decreased more rapidly in transgenic mice infected by isolate 19504lux from France than in the mice infected by isolate LNP14354lux from Africa, suggesting significantly higher virulence for the serogroup X isolate from Africa (Figure 2).
We next focused on differences between CC181 isolates obtained since 2006 and those obtained in the 1990s. We detected 18 genes for which alleles were identical in all isolates obtained since 2006 and in LNP14354 (obtained in the 1990s) but differed in LNP13407 (obtained in the 1990s), and we detected 23 genes for which alleles were identical in all isolates obtained since 2006 and in LNP13407 but differed in LNP14354 (Table 2).
Most of these 41 genes encode hypothetical proteins; several encode proteins that are involved in meningococcal survival in the blood (Table 2; Figure 1, panel C). In particular, genes of interest were fHbp (encodes factor H binding protein), lpt3 (encodes the phosphoethanolamine transferase), and lot3 (encodes the lipooligosaccharides, O-acetyltransferase). All serogroup X isolates obtained since the outbreak of 2006 (including those from the BIGSdb platform) harbored the lpt3 allele 45, which differs from allele 136 by the deletion of 2 codons (encoding Glu-211 and Ser-222 residues). These isolates do not react with the monoclonal antibody that recognizes immunotype L3,7,9 on the lipooligosaccharide (15). Replacing allele 45 with allele 136 by transformation enabled re-recognition of the epitope L3,7,9 (data not shown). The epitope L3,7,9 occurs frequently among invasive isolates, and antibodies against this epitope seem to be of importance for protection. Adding phosphoethanolamine on lipooligosaccharide may hide the epitope. This lipooligosaccharide modification may have enabled serogroup X to escape population immunity against lipooligosaccharides and contributed to its reemergence (15).
The extensive characterization of N. meningitidis serogroup X isolates in the meningitis belt of Africa supports the emergence of virulent serogroup X isolates and the belief that CC181 may be considered a hyperinvasive genetic lineage of meningococci. The occurrence of the periodic epidemic waves may be caused by the emergence and spread of successive new clones of meningococci with enhanced virulence and an ability to escape population immunity. Our data underline the need for whole-genome sequencing for reliable tracking of meningococcal isolates and emphasizes the need for an effective vaccine against serogroup X (5).
Dr. Agnememel is a microbiologist at the Institut Pasteur, Paris, and works on development of bacterial meningitis rapid diagnostic tools. His research interests include development of a universal vaccine against meningococcal meningitis.
Acknowledgment
This work was supported by the Institut Pasteur and by funding from the Fondation Total including a fellowship to A.A. This study made use of the Neisseria Multilocus Sequence Typing website (http://pubmlst.org/neisseria/), developed by Keith Jolley and sited at the University of Oxford, and the dynamic imaging facilities of Imagopole at the Institut Pasteur, Paris. The development of this site has been funded by the Wellcome Trust and European Union. This study also made use of the Meningitis Research Foundation Meningococcus Genome Library (http://www.meningitis.org/research/genome) developed by Public Health England, the Wellcome Trust Sanger Institute, and the University of Oxford.
References
- World Health Organization. Meningococcal disease control in countries of the African meningitis belt, 2013. Wkly Epidemiol Rec. 2014;89:206–14.PubMedGoogle Scholar
- Maiden MC, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A. 1998;95:3140–5. DOIPubMedGoogle Scholar
- Etienne J, Sperber G, Adamou A, Picq JJ. Epidemiological notes: meningococcal meningitis of serogroup X in Niamey (Niger) [in French]. Med Trop (Mars). 1990;50:227–9.PubMedGoogle Scholar
- Boisier P, Nicolas P, Djibo S, Taha MK, Jeanne I, Mainassara HB, Meningococcal meningitis: unprecedented incidence of serogroup X–related cases in 2006 in Niger. Clin Infect Dis. 2007;44:657–63. DOIPubMedGoogle Scholar
- Hong E, Giuliani MM, Deghmane AE, Comanducci M, Brunelli B, Dull P, Could the multicomponent meningococcal serogroup B vaccine (4CMenB) control Neisseria meningitidis capsular group X outbreaks in Africa? Vaccine. 2013;31:1113–6. DOIPubMedGoogle Scholar
- European Centre for Disease Prevention and Control. Annual epidemiological report 2012. Reporting on 2010 surveillance data and 2011 epidemic intelligence data. Stockholm: The Centre; 2013. p. 168–72.
- Maiden MC, van Rensburg MJ, Bray JE, Earle SG, Ford SA, Jolley KA, MLST revisited: the gene-by-gene approach to bacterial genomics. Nat Rev Microbiol. 2013;11:728–36. DOIPubMedGoogle Scholar
- Jolley KA, Hill DM, Bratcher HB, Harrison OB, Feavers IM, Parkhill J, Resolution of a meningococcal disease outbreak from whole-genome sequence data with rapid Web-based analysis methods. J Clin Microbiol. 2012;50:3046–53. DOIPubMedGoogle Scholar
- Kellogg DS Jr, Peacock WL Jr, Deacon WE, Brown L, Pirkle DI. Neisseria gonorrhoeae. I. Virulence genetically linked to clonal variation. J Bacteriol. 1963;85:1274–9.PubMedGoogle Scholar
- Veyrier FJ, Hong E, Deghmane AE, Taha MK. Draft genome sequence of a Neisseria meningitidis serogroup C isolate of sequence type 11 linked to an outbreak among men who have sex with men. Genome Announc. 2013;1:pii:e00795-13.
- Jolley KA, Maiden MC. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics. 2010;11:595. DOIPubMedGoogle Scholar
- Harrison OB, Bennett JS, Derrick JP, Maiden MC, Bayliss CD. Distribution and diversity of the haemoglobin–haptoglobin iron-acquisition systems in pathogenic and non-pathogenic Neisseria. Microbiology. 2013;159:1920–30. DOIPubMedGoogle Scholar
- Szatanik M, Hong E, Ruckly C, Ledroit M, Giorgini D, Jopek K, Experimental meningococcal sepsis in congenic transgenic mice expressing human transferrin. PLoS ONE. 2011;6:e22210. DOIPubMedGoogle Scholar
- Guiddir T, Deghmane AE, Giorgini D, Taha MK. Lipocalin 2 in cerebrospinal fluid as a marker of acute bacterial meningitis. BMC Infect Dis. 2014;14:276. DOIPubMedGoogle Scholar
- Rune Andersen S, Kolberg J, Hoiby EA, Namork E, Caugant DA, Oddvar Froholm L, Lipopolysaccharide heterogeneity and escape mechanisms of Neisseria meningitidis: possible consequences for vaccine development. Microb Pathog. 1997;23:139–55. DOIPubMedGoogle Scholar
Figures
Tables
Cite This Article1Current affiliation: Ministry of Health Panama and Instituto de Ciencias Médicas, Las Tablas, Panama.
Table of Contents – Volume 22, Number 4—April 2016
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Muhamed-Kheir Taha, Directeur Adjoint du Centre National de Reference des Meningocoques, Unite des Neisseria, Institut Pasteur, 28 Rue du Dr Roux, Paris 75724 CEDEX 15, France
Top