Volume 23, Number 4—April 2017
Dispatch
Detection and Molecular Characterization of Zoonotic Poxviruses Circulating in the Amazon Region of Colombia, 2014
Figure 2
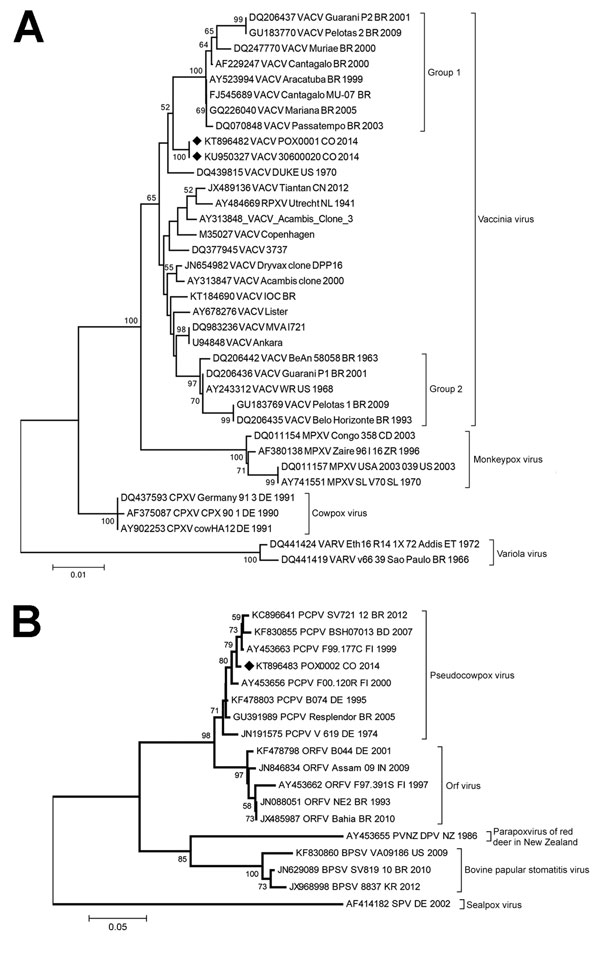
Figure 2. Phylogenetic characterization of orthopoxvirus gene A56R and parapoxvirus gene p37K of viruses obtained from patient lesion samples from an outbreak in Colombia, 2014. Trees were inferred by the neighbor-joining method. A) Nucleotide sequences of the A56R gene (829 bp) of reference orthopoxvirus strains were aligned and used for phylogenetic inference. The evolutionary distances were computed by using the T92+G model (shape: 0.69). Vaccinia virus (VACV) groups 1 and 2 are labeled with brackets. B) Nucleotide sequences of the partial (445 bp) p37K gene of reference parapoxvirus strains were aligned and used for phylogenetic inference. The evolutionary distances were computed by using the T92+G model (shape: 0.39). The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) are shown at the nodes. Diamonds indicate poxvirus isolates from Colombia. GenBank accession numbers and further information on the sequences included in the analyses are provided in Technical Appendix Tables 1, 2. Scale bars indicate nucleotide substitutions per site. BPSV, bovine papular stomatitis virus; CPXV, cowpox virus; RPXV, rabbitpox virus; MPXV, monkeypox virus; ORFV, orf virus; PCPXV, pseudocowpox virus; PVNZ, parapoxvirus of red deer in New Zealand; SPV, sealpox virus; VARV, variola virus.