Volume 23, Number 6—June 2017
Dispatch
Isolated Case of Marburg Virus Disease, Kampala, Uganda, 2014
Figure 2
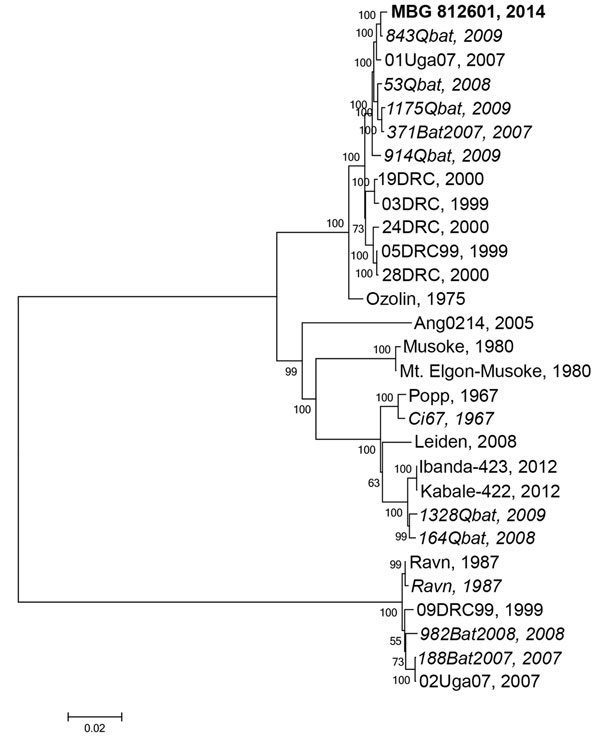
Figure 2. Phylogenetic tree comparing complete or nearly complete Marburg virus (MARV) genomes sequenced from bat and human sources in Uganda. A consensus whole-genome sequence was assembled by mapping reads to the reference MARV sequence NC_001608 using CLC Genomics Workbench (Waltham, MA, USA). A phylogenetic tree was constructed using MEGA6.06 (http://www.megasoftware.net). Viral sequences acquired from human sources are in standard type, and viral sequences acquired from bats are italicized; the sequence from the human case-patient described in this study, MBG 812601 2014, is in bold. Evolutionary history was inferred using the maximum-likelihood method based on the Tamura-Nei model with MEGA6.06. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. Values at nodes represent bootstrap values following 1,000 replicates. Scale bar represents substitutions per site. GenBank accession numbers used in this tree are KP985768, JX458855.1, FJ750957.1, JX458852.1, JX458854.1, FJ750958.1, JX458856.1, JX458828.1, JX458826.1, JX458834.1, DQ447651.1, JX458846.1, AY358025.2, DQ447657.1, Z12132.1, NC_001608.3, Z29337.1, EF446132.1, JN408064.1, KC545388.1, KC545387.1, DQ447649.1, EF446131.1, DQ447652.1, FJ750956.1, FJ750955.1, and FJ750953.1.