Volume 24, Number 4—April 2018
Dispatch
Genetic Characterization of Enterovirus A71 Circulating in Africa
Figure 1
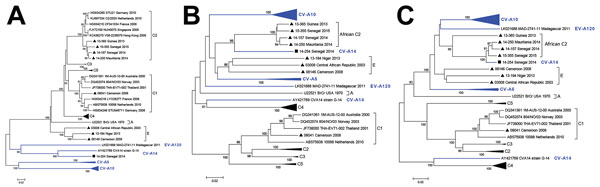
Figure 1. Phylogenetic relationships of the Africa enterovirus EV-A71 study strains based on A) P1 and B) P3 coding regions. An expanded version based on P1, P2, and P3 coding regions is online (LINK). Apart from the studied sequences, subgenomic datasets included their best nucleotide sequence matches identified by NCBI BLAST search (http://www.ncbi.nlm.nih.gov/BLAST) as well as representative sequences of different EV-A71 genogroups and subgenogroups originating worldwide. Trees were constructed from the nucleotide sequence alignment using MEGA 5.0. software (http://megasoftware.net/) with the neighbor-joining method. Distances were computed using the Kimura 2-parameter model. The robustness of the nodes was tested by 1,000 bootstrap replications. Bootstrap support values >75 are shown in nodes and indicate a strong support for the tree topology. For clarity, CV-A10, CV-A5, and EV-A71 subgenogroups C3, C4, and C5 have been collapsed. Study strains are indicated by laboratory code, country of origin, and year of isolation; previously published strains are indicated by GenBank accession number, isolate code, country of origin, and year of isolation. Black triangles indicate EV-A71 strains from this study; black square indicates the CV-A14 strain from this study. Strains gathered in brackets belong to EV-A71 genogroups or subgenogroups; strains marked in blue color belong to other species of EV-A. Scale bars indicate nucleotide substitutions per site. CV, coxsackievirus; EV, enterovirus.
1These first authors contributed equally to this article.
2These senior authors contributed equally to this article.