Volume 24, Number 7—July 2018
Synopsis
Typhus Group Rickettsiosis, Germany, 2010–20171
Cite This Article
Citation for Media
Abstract
Typhus group rickettsiosis is caused by the vectorborne bacteria Rickettsia typhi and R. prowazekii. R. typhi, which causes murine typhus, the less severe endemic form of typhus, is transmitted by fleas; R. prowazekii, which causes the severe epidemic form of typhus, is transmitted by body lice. To examine the immunology of human infection with typhus group rickettsiae, we retrospectively reviewed clinical signs and symptoms, laboratory changes, and travel destinations of 28 patients who had typhus group rickettsiosis diagnosed by the German Reference Center for Tropical Pathogens, Hamburg, Germany, during 2010–2017. Immunofluorescence assays of follow-up serum samples indicated simultaneous seroconversion of IgM, IgA, and IgG or concurrence in the first serum sample. Cytokine levels peaked during the second week of infection, coinciding with organ dysfunction and seroconversion. For 3 patients, R. typhi was detected by species-specific nested quantitative PCR. For all 28 patients, R. typhi was the most likely causative pathogen.
Typhus group rickettsiosis (TGR) is caused by Rickettsia typhi and R. prowazekii, 2 Biosafety Level 3 organisms of the family Rickettsiaceae, which comprises obligate intracellular gram-negative zoonotic bacteria. R. typhi is responsible for murine typhus, the endemic fleaborne form of typhus, which is emerging in predominantly tropical coastal regions. R. prowazekii is responsible for epidemic louseborne typhus in temperate and tropical regions. R. prowazekii is classified as a Centers for Disease Control and Prevention category B bioweapon pathogen.
The clinical picture of TGR caused by either pathogen is similar: fever, headache, and exanthema (1,2). Inoculation eschars, which are classically seen in patients with spotted fever group rickettsioses (SFGR), are usually absent in patients with TGR. Cardiac, pulmonary, and central nervous system (CNS) complications can occur during the course of infection (1,2). R. typhi infection is usually milder than R. prowazekii infection. Fatality rates among patients with untreated typhus are ≈4% (3,4) among patients with R. typhi infection and 13%–30% (2) among those with R. prowazekii infection.
To learn more about the immunology of human infection with typhus group rickettsiae, we retrospectively analyzed TGR cases diagnosed at the National Reference Center for Tropical Pathogens in Hamburg, Germany, during 2010–2017. We collected clinical data and analyzed patient serum. Antibody kinetics were determined from follow-up serum samples. Serum cytokine responses were measured by flow cytometry from all available serum samples. In addition, we used novel nested quantitative PCRs targeting the prsA genes of R. typhi and R. prowazekii on archived clinical material.
Cases and Inclusion Criteria
We screened the database of the German Reference Center for Tropical Pathogens at the Bernhard Nocht Institute for Tropical Medicine in Hamburg for autochthonous and imported (travel- or migration-associated) TGR cases diagnosed from January 1, 2010, through December 31, 2017. Written general consent had been obtained from patients before the study. TGR cases were defined as a clinically compatible disease with >1 of the following laboratory test results: a positive PCR and sequencing result, seroconversion to TGR antigens in an indirect immunofluorescence assay (IFA), parallel TGR IgM and IgG detection in a single sample by IFA, or a single IFA IgG or total Ig titer of >320. In addition, antibodies against SFGR antigens, when available, had to be lower than TGR antigens in the IFA. Serologic testing results for leptospirosis (in-house ELISA), scrub typhus (in-house IFA), and dengue fever (in-house IFA) had to be negative.
Serologic Assays
We performed in-house TGR IFA by using R. typhi strain Wilmington and R. prowazekii strain Madrid E grown in L929 mouse fibroblast cell culture. IFA reference values for R. typhi and R. prowazekii were <40 (IgM), <20 (IgA), and <80 (IgG and total Ig). In parallel, we performed in-house SFGR IFA with R. conorii strain 7 (ATCC VR-613) by using the same culture conditions and with reference values of <20 (IgM and IgA) and <40 (IgG and total Ig). All reference values were determined with serum from 200 healthy Caucasian blood donors.
Molecular Assays
We performed panrickettsial real-time quantitative PCR (qPCR) targeting the ompB gene (5). A nested qPCR detecting specifically the prsA gene of R. typhi (6) was used, and a nested qPCR for amplifying the prsA gene of R. prowazekii was developed by using outer primers GCTTGCAGAAGAATTCTCTCTTG (forward) and GGCACAGGTTTTTTTTCAAGCAC (reverse) and nested primers CAGCGTCAAATGGTGGGATT (forward) and TGCCAACCGAAACTTGTTTTG (reverse) with established cycling conditions (6). Probes were 6FAM-ATCAATCAGGGCAATTAGTACCAGAA-BHQ1 for R. typhi and 6FAM-ATCAACCAGGGCAGTTAGTACCAGAA-BHQ1 for R. prowazekii. We used conventional gel PCRs for later sequencing, followed by BLAST analysis (http://www.blast.ncbi.nlm.nih.gov). We performed PCRs from DNA extracts from blood in EDTA, from the first archived serum sample if available, and in 1 case from a formalin-fixed paraffin-embedded liver biopsy sample.
Cytokine Measurements
For all available serum samples, we analyzed serum cytokine responses by using LEGENDplex (BioLegend, Fell, Germany). For controls, we used 16 serum samples from healthy blood donors. For cytokine analysis, we assigned blood sampling dates from the patients as acute phase of infection (days 1–7 and days 8–14 of illness), prolonged phase (days 15–28 of illness), and convalescent phase (days 29–56 of illness).
Figure 1
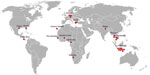
Figure 1. Countries and US states in which 27 of 28 patients acquired typhus group rickettsiosis diagnosed in Germany, 2010–2017. For 1 of the 28 patients, no information was available. Most infections were...
We identified 28 TGR patients (Table 1); age range was 4–80 years (mean age 38.3 years), and male:female ratio was 1.5:1. For 1 patient, no information about travel destination or medical history was available; only age and sex information was available. TGR infections had been acquired during travel (Figure 1), primarily to Southeast Asia (Indonesia, Thailand, Cambodia, [13 (46%) cases]); most infections were acquired in Indonesia (8 [29%] cases). Other cases were acquired in Europe (Germany, Greece, Canary Islands; 6 [21%] cases), Africa (The Gambia, Burkina Faso, Cameroon, Namibia; 4 [14%] cases), North America (Florida and Texas, USA; 2 [7%] cases), Costa Rica (1 case), and Nepal (1 case). Three infections were locally acquired in Germany by patients without a travel history (patients 9, 15, and 21). No patients had been asked about exposure to rats.
Figure 2

Figure 2. Typical exanthema in a typhus patient after travel to Thailand. The rash is maculopapular and nonpruritic.
Of note, patients were examined on different days of illness at different hospitals. At the time of initial examination, the most frequently reported sign or symptom was fever (79%), followed by exanthema (50%; Figure 2), headache (46%), myalgia/arthralgia (25%), cough/pneumonia (15%), and splenomegaly (11%). Only 6 (21%) patients had the classical TGR triad of fever, headache, and exanthema. The following were recorded in the medical records of 1 (4%) patient each: neurologic signs, lymphadenopathy, herpes simplex reactivation, acute kidney injury, dysuria, diarrhea, and ear redness. Hospitalization was necessary for 18 (64%) patients. Patients had received doxycycline (200 mg/d for 5–14 d) in the country of travel or after return; all recovered from infection without sequelae.
At the time of initial examination, laboratory changes were reported in the medical records of 23 patients and included increased levels of C-reactive protein (70%), liver enzymes (65%), lactate dehydrogenase (LDH, 13%), and creatine kinase (13%); thrombocytopenia (26%); anemia (17%); and leukocytosis (17%). A positive PCR for R. typhi was obtained for 3 (10.7%) patients: on days 7 and 10 of infection from whole blood by panrickettsial ompB-qPCR and R. typhi–specific nested prsA-qPCR (patients 28 and 17), and on day 10 from a liver biopsy sample by R. typhi–specific nested prsA-qPCR only (patient 13). We confirmed identity of R. typhi by sequencing of the 856 bp ompB gene fragment, which showed 100% identity to GenBank entries of R. typhi clone 4, strain B9991CWPP, strain TH1527, and strain Wilmington (GenBank accession nos. KF241858, CP003398, CP003397, and AE017197). Sequencing of the 140 bp prsA gene fragment showed 98% identity to the same strains. No positive PCR for R. prowazekii was obtained (Table 1).
The earliest that antibodies against TGR antigens were detected was day 7 of illness. The median day for seroconversion was day 12. The percentages of seroconversion and parallel and single antibody class detection against TGR antigens are shown in Table 2. For 5 patients, only limited serologic information was available because a limited quantity of serum had been stored for retesting. Antibody titers of any class varied among patients despite illness onset occurring on the same day. TGR-specific IgA titers were identical or lower and even undetectable for some patients but never higher than TGR-specific IgM titers except for 1 patient for whom specific IgA and IgG but not IgM were detected. No serologic differentiation between R. typhi and R. prowazekii was achieved by IFA; titer differences between the 2 species for all antibody classes were <2 for all patients.
Figure 3

Figure 3. Cytokine and chemokine levels in serum from patients with imported and autochthonous typhus group rickettsiosis and controls, Germany, 2010–2017. Using a bead-based LEGENDplex assay (BioLegend, Fell, Germany), we analyzed 16 serum...
Serum cytokines (Figure 3) could be measured for 21 (75%) patients; for 11 patients, they were measured as kinetics at different time points (2–5 time points). Illness was determined to be in the acute phase according to 17 serum samples (7 on days 0–7 and 10 on days 8–14), in the prolonged phase for 4 samples, and in the convalescent phase for 5. Concentrations of interferon γ–induced protein (IP) 10 and vascular endothelial growth factor (VEGF) were elevated in the first week of the acute phase, peaked during the second week, and then declined. This trend was also observed for interleukin (IL) 8, except for patient 26, for whom IL-8 increased continuously (from 101 pg/mL on day 5 of illness and 245 pg/mL on day 12 up to 12,380 pg/mL on day 29). Serum levels of interferon-γ, IL-1β, IL-6, IL-8, IP-10, macrophage inflammatory proteins 1α and 1β, and VEGF were substantially increased over those in healthy controls in the second week of the acute phase of illness. The serum concentrations of IL-21 and IL-22 started to elevate during the second week of the acute phase, peaked during the prolonged phase, and were not detectable in the convalescent phase. Serum levels of interferon-α were substantially increased during the second and third weeks of illness; IL-10 was also elevated during this time. The serum concentrations of the following were comparable between controls and patients at all analyzed time points (data not shown): basic fibroblast growth factor, eotaxin, granulocyte colony stimulating factor, granulocyte-macrophage colony stimulating factor, IL-2, IL-4, IL-5, IL-9, IL-12p70, IL-13, IL-17A, IL-17F, monocyte chemotactic protein (MCP) 1, platelet-derived growth factor, RANTES (regulated on activation, normal T cell expressed and secreted), and tumor necrosis factor α.
The rather benign course of illness and outcomes, the travel destinations, and the molecular identification of R. typhi for a few patients indicate that murine (endemic) typhus is the most likely diagnosis for all TGR patients in our study. We found no cases of definitive R. prowazekii infection (i.e., epidemic typhus). IFA testing did not allow for TGR species discrimination because no significant titer differences were noted between serum tested for R. typhi and R. prowazekii.
As expected, most TGR cases diagnosed in our study were travel associated. Nearly half of the infections were acquired in Southeast Asia. A surveillance study also found Southeast Asia to be the most common region of exposure for patients with murine typhus (7). Another study found Southeast Asia to rank second after Africa (8). We identified 3 autochthonous cases in patients from Germany, but the circumstances of infection and the exposure (rats or flea bites) could not be identified. For these 3 patients (50, 72, and 80 years of age), we could not exclude the possibility of a relapse of R. prowazekii infection (Brill-Zinsser disease) acquired during World War II or in early postwar Germany. For Europe, exposure to R. typhi had been previously recorded in Spain (9); Canary Islands and Greece (8); and Cyprus, Italy, France, Croatia and Slovenia (10).
The clinical signs of illness found in our study were undifferentiated, except for a maculopapular rash that had developed for half of the patients. The typical triad for murine typhus (fever, headache, and exanthema) occurred in less than one fourth of the patients reported here. According to similar findings by a study of comparable size in France, the triad was considered nonspecific (8); the triad occurred in one third of patients according to a study from Texas (11) and a recent review by others (1). In our study, nearly two thirds of the patients were hospitalized during travel or after return home. A similar high rate of 60% for TGR-associated hospitalization in Texas has been described (11). Complications, mostly pulmonary or CNS, have reached 26% among patients with murine typhus (1). In our study, the complication rate was 18%; complications included pneumonia, acute kidney injury, and CNS involvement.
In our study, laboratory data, which could not be retrieved in detail for all patients, often showed elevated levels of C-reactive protein, LDH, and liver enzymes, paralleled by thrombocytopenia. More than 70% of patients with murine typhus had increased liver enzyme and LDH levels, and nearly half had thrombocytopenia, as described in a recent review (1). Because many patients in our study had fever, exanthema, elevated liver enzymes, and thrombocytopenia, the differential diagnoses for travelers, especially to Asia, include scrub typhus, dengue fever, and leptospirosis. All patients in our study were negative for these infections. Thus, infection with R. typhi should be considered for patients with fever, headache, exanthema, and concurrent thrombocytopenia and elevated levels of liver enzymes, particularly patients who have recently traveled to Southeast Asia.
In our study, molecular detection of TGR species was positive for a liver biopsy and 2 whole blood samples but negative for archived serum. This finding is in line with the higher sensitivity of rickettsial PCR from whole blood or buffy coat than from serum (12).
Typically, diagnostic IgM and IgG are simultaneously detected 7–15 days after onset of symptoms (13) and titers are detected by IFA for 50% of patients at the end of the first week of symptoms and for nearly all after 2 weeks (4). In our study, the earliest detection of antibodies against TGR was day 7 of illness, and the median day of seroconversion was day 12. Seroconversion, which is delayed in patients with TGR rickettsiosis, involved IgM, IgA, and IgG simultaneously. In nearly half of the patients in our study, antibodies of these 3 classes were detected in parallel against TGR in the first serum sample collected. It remains tempting to speculate about an initial depression of B-cell responses by TGR rickettsiae. IgA testing had been included in the study to test for possible earlier seroconversion with antibodies in this class, but IgA detection proved less sensitive than IgM detection. Of note, because serum had not been collected from all patients on the same day and was not collected every day, the time points can only serve as estimates.
We report data on the systemic inflammatory response in patients with TGR during 8 weeks after illness onset. Data on circulating levels of inflammatory mediators are available for only a few SFGR diseases, probably because of the limited availability of patient samples. Most cytokine and chemokine elevations in TGR patients in our study occurred in the second week of infection, coinciding with organ dysfunction and seroconversion. The mechanisms expected to contribute to the vascular permeability observed in patients with clinical disease include the effects of inflammatory cells and their mediators (14). Of note, vascular dysfunction and damage in the infected host most likely contribute to the pathogenesis of human rickettsial diseases. Because endothelial cells are the major target cells for rickettsial infections (15) and have emerged as key immune-reactive cells involved in host defense and inflammation (16,17), prior studies have focused on the behavior and inflammatory phenotype of these cells after infection in vitro. Endothelial cells react to infection with SFGR species by increased expression of cytokines such as IL-1 and IL-6 and chemokines such as IL-8 and MCP-1, which favor the migration of leukocytes (18–20). Experiments with cultured endothelial cells showed that they also become activated by R. prowazekii, which induces the expression of proinflammatory cytokines and chemokines (21). Endothelial cells upregulate the expression of the cytokines tumor necrosis factor-α, IL-1α, and IL-6 and the chemokines IP-10, MCP-1, and RANTES, which leads to transmigration of peripheral blood mononuclear cells that acquire an inflammatory transcriptional profile. Infection of endothelial cells with R. typhi resulted in enhanced expression of IP-10, MCP-1, and RANTES in infected endothelial cells; the expression of IL-8 was also upregulated (22). Although different intracytoplasmic behavior of SFGR and TGR pathogens has been shown (22–25), these findings indicate that endothelial cells react in a comparable way after infection with pathogens of either group. In fact, the elevations of the chemokines IL-8, IP-10, macrophage inflammatory protein 1α, IL-6, and IL-10 that we found in the serum of the patients in our study during the acute phase of illness were also detectable in patients with SFGR, such as African tick bite fever caused by R. africae and Mediterranean spotted fever caused by R. conorii (26–28). Moreover, inflammatory cytokines such as IL-1β, IL-6, and IL-8 upregulate VEGF expression (29). Of note, the serum concentrations of all these mediators were substantially elevated during the same period in the patients in our study (precisely in the second week of illness). VEGF and IL-8 are important mediators of angiogenesis and might contribute to initiation of repair mechanisms after endothelial damage through rickettsial growth and spread (30). In addition, IL-8 induces neutrophil mobilization and activation (31). Neutrophils are the first cells of the innate immune system that migrate to the site of infection and participate in bacterial defense.
In our study, interferon-γ levels were substantially elevated in the serum of patients in the acute phase of disease and seem to play a crucial role in antirickettsial immune responses. Interferon-γ has protective features in host defense during infection of susceptible mouse strains with SFGR and TGR species (32–37), and it adversely affects the growth of TGR species in various host cells (36–39). Therefore, the early interferon-γ response could activate intracellular bactericidal mechanisms to further control the spread of infection.
The concentrations of IL-21 and IL-22 in the serum of TGR patients started to increase in the second week of illness, peaked in the third week, and were no longer detectable 1 month after symptom onset. IL-22 seems to have protective functions because it increases the production of neutrophilic granulocyte-attracting chemokines such as IL-8, protects tissues from damage, and enhances tissue regeneration (40,41). IL-22–responsive cells are distributed throughout the body in several organs including those from the digestive (pancreas, liver, colon) and respiratory (lung, trachea) systems and the skin (42). Whether endothelial cells express the IL-22 receptor complex remains to be determined. IL-22 is produced by several types of cells of the lymphoid lineage and include activated T-cells as well as innate lymphoid cells such as natural killer cells, lymphoid tissue inducer cells, and lymphoid tissue inducer–like cells (43–50). Which cell types are responsible for IL-22 secretion and the protective potential of this cytokine during rickettsial infections remains to be elucidated.
In conclusion, our data broaden the knowledge of TGR immunology and diagnosis and shed light on immunologic changes that occur during successive weeks of illness. However, more investigations of immunologic changes, including analyses of human B and T cells, are needed.
Dr. Rauch is a biochemist at the National Reference Center for Tropical Diseases, Bernhard Nocht Institute, Hamburg, Germany. Her research interests focus on the development of diagnostic methods and the immune responses in rickettsial diseases.
References
- Tsioutis C, Zafeiri M, Avramopoulos A, Prousali E, Miligkos M, Karageorgos SA. Clinical and laboratory characteristics, epidemiology, and outcomes of murine typhus: A systematic review. Acta Trop. 2017;166:16–24. DOIPubMedGoogle Scholar
- Angelakis E, Bechah Y, Raoult D. The history of epidemic typhus. Microbiol Spectr. 2016;4:4.PubMedGoogle Scholar
- Raoult D, Roux V. Rickettsioses as paradigms of new or emerging infectious diseases. Clin Microbiol Rev. 1997;10:694–719.PubMedGoogle Scholar
- Dumler JS, Taylor JP, Walker DH. Clinical and laboratory features of murine typhus in south Texas, 1980 through 1987. JAMA. 1991;266:1365–70. DOIPubMedGoogle Scholar
- Keller C, Krüger A, Schwarz NG, Rakotozandrindrainy R, Rakotondrainiarivelo JP, Razafindrabe T, et al. High detection rate of Rickettsia africae in Amblyomma variegatum but low prevalence of anti-rickettsial antibodies in healthy pregnant women in Madagascar. Ticks Tick Borne Dis. 2016;7:60–5. DOIPubMedGoogle Scholar
- Papp S, Rauch J, Kuehl S, Richardt U, Keller C, Osterloh A. Comparative evaluation of two Rickettsia typhi-specific quantitative real-time PCRs for research and diagnostic purposes. Med Microbiol Immunol (Berl). 2017;206:41–51. DOIPubMedGoogle Scholar
- Jensenius M, Davis X, von Sonnenburg F, Schwartz E, Keystone JS, Leder K, et al.; GeoSentinel Surveillance Network. Multicenter GeoSentinel analysis of rickettsial diseases in international travelers, 1996-2008. Emerg Infect Dis. 2009;15:1791–8. DOIPubMedGoogle Scholar
- Walter G, Botelho-Nevers E, Socolovschi C, Raoult D, Parola P. Murine typhus in returned travelers: a report of thirty-two cases. Am J Trop Med Hyg. 2012;86:1049–53. DOIPubMedGoogle Scholar
- Nogueras MM, Cardeñosa N, Sanfeliu I, Muñoz T, Font B, Segura F. Evidence of infection in humans with Rickettsia typhi and Rickettsia felis in Catalonia in the Northeast of Spain. Ann N Y Acad Sci. 2006;1078:159–61. DOIPubMedGoogle Scholar
- Angelakis E, Botelho E, Socolovschi C, Sobas CR, Piketty C, Parola P, et al. Murine typhus as a cause of Fever in travelers from Tunisia and mediterranean areas. J Travel Med. 2010;17:310–5. DOIPubMedGoogle Scholar
- Murray KO, Evert N, Mayes B, Fonken E, Erickson T, Garcia MN, et al. Typhus group rickettsiosis, Texas, USA, 2003–2013. Emerg Infect Dis. 2017;23:645–8. DOIPubMedGoogle Scholar
- Watthanaworawit W, Turner P, Turner C, Tanganuchitcharnchai A, Richards AL, Bourzac KM, et al. A prospective evaluation of real-time PCR assays for the detection of Orientia tsutsugamushi and Rickettsia spp. for early diagnosis of rickettsial infections during the acute phase of undifferentiated febrile illness. Am J Trop Med Hyg. 2013;89:308–10. DOIPubMedGoogle Scholar
- Brouqui P, Bacellar F, Baranton G, Birtles RJ, Bjoërsdorff A, Blanco JR, et al.; ESCMID Study Group on Coxiella, Anaplasma, Rickettsia and Bartonella; European Network for Surveillance of Tick-Borne Diseases. Guidelines for the diagnosis of tick-borne bacterial diseases in Europe. Clin Microbiol Infect. 2004;10:1108–32. DOIPubMedGoogle Scholar
- Mansueto P, Vitale G, Di Lorenzo G, Arcoleo F, Mansueto S, Cillari E. Immunology of human rickettsial diseases. J Biol Regul Homeost Agents. 2008;22:131–9.PubMedGoogle Scholar
- Sahni SK, Rydkina E. Host-cell interactions with pathogenic Rickettsia species. Future Microbiol. 2009;4:323–39. DOIPubMedGoogle Scholar
- Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7:803–15. DOIPubMedGoogle Scholar
- Sahni SK. Endothelial cell infection and hemostasis. Thromb Res. 2007;119:531–49. DOIPubMedGoogle Scholar
- Clifton DR, Rydkina E, Huyck H, Pryhuber G, Freeman RS, Silverman DJ, et al. Expression and secretion of chemotactic cytokines IL-8 and MCP-1 by human endothelial cells after Rickettsia rickettsii infection: regulation by nuclear transcription factor NF-kappaB. Int J Med Microbiol. 2005;295:267–78. DOIPubMedGoogle Scholar
- Kaplanski G, Teysseire N, Farnarier C, Kaplanski S, Lissitzky JC, Durand JM, et al. IL-6 and IL-8 production from cultured human endothelial cells stimulated by infection with Rickettsia conorii via a cell-associated IL-1 alpha-dependent pathway. J Clin Invest. 1995;96:2839–44. DOIPubMedGoogle Scholar
- Sporn LA, Marder VJ. Interleukin-1 alpha production during Rickettsia rickettsii infection of cultured endothelial cells: potential role in autocrine cell stimulation. Infect Immun. 1996;64:1609–13.PubMedGoogle Scholar
- Bechah Y, Capo C, Raoult D, Mege JL. Infection of endothelial cells with virulent Rickettsia prowazekii increases the transmigration of leukocytes. J Infect Dis. 2008;197:142–7. DOIPubMedGoogle Scholar
- Rydkina E, Sahni A, Silverman DJ, Sahni SK. Comparative analysis of host-cell signalling mechanisms activated in response to infection with Rickettsia conorii and Rickettsia typhi. J Med Microbiol. 2007;56:896–906. DOIPubMedGoogle Scholar
- Gouin E, Gantelet H, Egile C, Lasa I, Ohayon H, Villiers V, et al. A comparative study of the actin-based motilities of the pathogenic bacteria Listeria monocytogenes, Shigella flexneri and Rickettsia conorii. J Cell Sci. 1999;112:1697–708.PubMedGoogle Scholar
- Heinzen RA. Rickettsial actin-based motility: behavior and involvement of cytoskeletal regulators. Ann N Y Acad Sci. 2003;990:535–47. DOIPubMedGoogle Scholar
- Heinzen RA, Grieshaber SS, Van Kirk LS, Devin CJ. Dynamics of actin-based movement by Rickettsia rickettsii in vero cells. Infect Immun. 1999;67:4201–7.PubMedGoogle Scholar
- Damås JK, Davì G, Jensenius M, Santilli F, Otterdal K, Ueland T, et al. Relative chemokine and adhesion molecule expression in Mediterranean spotted fever and African tick bite fever. J Infect. 2009;58:68–75. DOIPubMedGoogle Scholar
- Jensenius M, Ueland T, Fournier PE, Brosstad F, Stylianou E, Vene S, et al. Systemic inflammatory responses in African tick-bite fever. J Infect Dis. 2003;187:1332–6. DOIPubMedGoogle Scholar
- Vitale G, Mansueto S, Gambino G, Mocciaro C, Spinelli A, Rini GB, et al. The acute phase response in Sicilian patients with boutonneuse fever admitted to hospitals in Palermo, 1992-1997. J Infect. 2001;42:33–9. DOIPubMedGoogle Scholar
- Angelo LS, Kurzrock R. Vascular endothelial growth factor and its relationship to inflammatory mediators. Clin Cancer Res. 2007;13:2825–30. DOIPubMedGoogle Scholar
- Valbuena G, Walker DH. Infection of the endothelium by members of the order Rickettsiales. Thromb Haemost. 2009;102:1071–9. DOIPubMedGoogle Scholar
- Harada A, Sekido N, Akahoshi T, Wada T, Mukaida N, Matsushima K. Essential involvement of interleukin-8 (IL-8) in acute inflammation. J Leukoc Biol. 1994;56:559–64. DOIPubMedGoogle Scholar
- Walker DH, Popov VL, Feng HM. Establishment of a novel endothelial target mouse model of a typhus group rickettsiosis: evidence for critical roles for gamma interferon and CD8 T lymphocytes. Lab Invest. 2000;80:1361–72. DOIPubMedGoogle Scholar
- Li H, Jerrells TR, Spitalny GL, Walker DH. Gamma interferon as a crucial host defense against Rickettsia conorii in vivo. Infect Immun. 1987;55:1252–5.PubMedGoogle Scholar
- Walker DH, Olano JP, Feng HM. Critical role of cytotoxic T lymphocytes in immune clearance of rickettsial infection. Infect Immun. 2001;69:1841–6. DOIPubMedGoogle Scholar
- Feng HM, Popov VL, Walker DH. Depletion of gamma interferon and tumor necrosis factor alpha in mice with Rickettsia conorii-infected endothelium: impairment of rickettsicidal nitric oxide production resulting in fatal, overwhelming rickettsial disease. Infect Immun. 1994;62:1952–60.PubMedGoogle Scholar
- Moderzynski K, Heine L, Rauch J, Papp S, Kuehl S, Richardt U, et al. Cytotoxic effector functions of T cells are not required for protective immunity against fatal Rickettsia typhi infection in a murine model of infection: Role of TH1 and TH17 cytokines in protection and pathology. PLoS Negl Trop Dis. 2017;11:e0005404. DOIPubMedGoogle Scholar
- Moderzynski K, Papp S, Rauch J, Heine L, Kuehl S, Richardt U, et al. CD4+ T cells are as protective as CD8+ T cells against Rickettsia typhi infection by activating macrophage bactericidal activity. PLoS Negl Trop Dis. 2016;10:e0005089. DOIPubMedGoogle Scholar
- Turco J, Winkler HH. Effect of mouse lymphokines and cloned mouse interferon-gamma on the interaction of Rickettsia prowazekii with mouse macrophage-like RAW264.7 cells. Infect Immun. 1984;45:303–8.PubMedGoogle Scholar
- Turco J, Winkler HH. Gamma-interferon-induced inhibition of the growth of Rickettsia prowazekii in fibroblasts cannot be explained by the degradation of tryptophan or other amino acids. Infect Immun. 1986;53:38–46.PubMedGoogle Scholar
- Boniface K, Bernard FX, Garcia M, Gurney AL, Lecron JC, Morel F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol. 2005;174:3695–702. DOIPubMedGoogle Scholar
- Wolk K, Haugen HS, Xu W, Witte E, Waggie K, Anderson M, et al. IL-22 and IL-20 are key mediators of the epidermal alterations in psoriasis while IL-17 and IFN-gamma are not. J Mol Med (Berl). 2009;87:523–36. DOIPubMedGoogle Scholar
- Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity. 2004;21:241–54. DOIPubMedGoogle Scholar
- Crellin NK, Trifari S, Kaplan CD, Cupedo T, Spits H. Human NKp44+IL-22+ cells and LTi-like cells constitute a stable RORC+ lineage distinct from conventional natural killer cells. J Exp Med. 2010;207:281–90. DOIPubMedGoogle Scholar
- Hughes T, Becknell B, Freud AG, McClory S, Briercheck E, Yu J, et al. Interleukin-1beta selectively expands and sustains interleukin-22+ immature human natural killer cells in secondary lymphoid tissue. Immunity. 2010;32:803–14. DOIPubMedGoogle Scholar
- Kim MS, Kim WS, Piao ZH, Yun S, Lee SH, Lee S, et al. IL-22 producing NKp46+ innate lymphoid cells can differentiate from hematopoietic precursor cells. Immunol Lett. 2011;141:61–7. DOIPubMedGoogle Scholar
- Kim S, Han S, Withers DR, Gaspal F, Bae J, Baik S, et al. CD117+ CD3− CD56− OX40Lhigh cells express IL-22 and display an LTi phenotype in human secondary lymphoid tissues. Eur J Immunol. 2011;41:1563–72. DOIPubMedGoogle Scholar
- Ness-Schwickerath KJ, Jin C, Morita CT. Cytokine requirements for the differentiation and expansion of IL-17A- and IL-22-producing human Vgamma2Vdelta2 T cells. J Immunol. 2010;184:7268–80. DOIPubMedGoogle Scholar
- Nograles KE, Zaba LC, Shemer A, Fuentes-Duculan J, Cardinale I, Kikuchi T, et al. IL-22-producing “T22” T cells account for upregulated IL-22 in atopic dermatitis despite reduced IL-17-producing TH17 T cells. J Allergy Clin Immunol. 2009;123:1244–52 e2.
- Ortega C, Fernández-A S, Carrillo JM, Romero P, Molina IJ, Moreno JC, et al. IL-17-producing CD8+ T lymphocytes from psoriasis skin plaques are cytotoxic effector cells that secrete Th17-related cytokines. J Leukoc Biol. 2009;86:435–43. DOIPubMedGoogle Scholar
- Wolk K, Kunz S, Asadullah K, Sabat R. Cutting edge: immune cells as sources and targets of the IL-10 family members? J Immunol. 2002;168:5397–402. DOIPubMedGoogle Scholar
Figures
Tables
Cite This Article1Part of this study was orally presented at the annual meeting of the German Society for Hygiene and Microbiology, February 19–21, 2018, Bochum, Germany.
Table of Contents – Volume 24, Number 7—July 2018
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Dennis Tappe, Bernhard Nocht Institute for Tropical Medicine, Bernhard-Nocht-Str. 74, 20359 Hamburg, Germany
Top