Volume 25, Number 2—February 2019
Dispatch
Bat Influenza A(HL18NL11) Virus in Fruit Bats, Brazil
Cite This Article
Citation for Media
Abstract
Screening of 533 bats for influenza A viruses showed subtype HL18NL11 in intestines of 2 great fruit-eating bats (Artibeus lituratus). High concentrations suggested fecal shedding. Genomic characterizations revealed conservation of viral genes across different host species, countries, and sampling years, suggesting a conserved cellular receptor and wide-ranging occurrence of bat influenza A viruses.
Influenza A viruses are major causes of human disease and are predominantly maintained in avian reservoirs (1). The segmented influenza A genome facilitates reassortment events in birds or intermediate hosts, such as swine and horses, leading to emergence of new variants potentially capable of causing zoonotic infections (2). Bats are major sources of zoonotic pathogens (3). In pioneering studies from 2012 and 2013, the first bat influenza A viruses, termed H17N10 and H18N11, were discovered in 2 bat species, Sturnira lilium (little yellow-shouldered bat) and Artibeus planirostris (flat-faced fruit-eating bat) (4,5).
Bat-associated influenza A viruses are phylogenetically highly divergent from avian-associated influenza A viruses in their hemagglutinin (HA) and neuraminidase (NA) genes, suggesting these viruses represent ancient influenza A strains (2). Consistent with their genetic divergence, bat-associated influenza A surface proteins lack typical hemagglutination and neuraminidase activities (6), leading to the terminology HA-like (HL) and neuraminidase-like (NL) for bat-associated influenza surface proteins.
So far, only 4 individual bat specimens yielded influenza A genomic sequences during the pivotal investigations (4,5). HL18NL11 has only been found in 1 A. planirostris bat captured in Peru in 2010 (5), challenging definite host assessments. To investigate bat influenza A virus epidemiology, we investigated bats in southern Brazil during 2010–2014.
For this study, we sampled 533 individual bats representing 26 species and 3 families across 28 sampling sites (Table 1). Bats were captured using mist nets, euthanized, and necropsied and were identified on the basis of morphological criteria by trained field biologists as described previously (7). Only intestine samples were available for virological analyses. The Instituto Brasileiro do Meio Ambiente e dos Recursos Naturais (21748–1), Instituto Ambiental do Paraná (235/10), and the ethics committee of the Institute of Biomedical Science from the University of São Paulo (56–18–03/2014) authorized sampling.
Figure 1
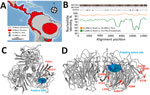
Figure 1. Bat influenza A(HL18NL11) virus detection and genomic characterization, Brazil, 2010–2014. A) Distribution of Artibeus species bats carrying HL18NL11 in Central and South America, according to the Red List of Threatened Species...
We tested intestine specimens from all bats using 2 highly sensitive, broadly reactive nested reverse transcription PCRs targeting different regions of the influenza A polymerase basic (PB) 1 gene (5,8). Positive results on both tests came from only 2 samples, from Artibeus lituratus great fruit-eating bats captured on March 7 and March 12, 2012, at 2 locations separated by 12 km in an Atlantic rainforest patch. No other sample was positive, yielding a 10.0% (2/20) overall detection rate in this site and 16.7% (2/12) detection in A. lituratus bats from this site (Table 1; Figure 1, panel A). Neither bat testing positive for influenza A virus showed signs of disease. A. lituratus bats were the most abundantly sampled species (Table 1).
The low overall influenza virus detection rate in this study (0.4%, 95% CI 0.0%–1.5%) was not significantly different by Fisher exact test from the previous 2 studies (1/110 bats for HL18NL11 [0.9%, 95% CI 0.0%–5.5%; p = 0.43]; 3/316 bats for HL17NL10 [1.0%, 95% CI 0.0%–2.9%; p = 0.37]). Apparently low rates of acute influenza A virus infection in bats are not consistent with high seroprevalence of 72% in different bat species according to a preliminary investigation (5) and may hint at seasonal variation in bat influenza virus infections, comparable to other batborne RNA viruses (9).
Sanger sequencing of the screening PCR amplicons suggested close genetic relatedness of the strains circulating in Brazil with the HL18NL11 strain circulating in Peru. Virus concentrations in the positive intestine specimens as determined by strain-specific quantitative real-time reverse transcription RT-PCR (Appendix Table 1) were high (1.5 × 109 and 4.9 × 1010 RNA copies/g of tissue). High HL18NL11 concentrations in intestinal specimens are consistent with qualitative data from the pioneering study on HL18NL11 (5) and may suggest intestinal tropism and potential fecal shedding into the environment.
We determined the full coding sequence of all 8 segments of the viral genomes using primers aiming at amplifying overlapping regions of bat influenza A virus genomes (GenBank accession nos. MH682200–15) (Appendix Table 1). The 2 HL18NL11 variants in Brazil differed by 15 nt from each other across the combined 8 genomic segments. Four of those substitutions were nonsynonymous, causing amino acid exchanges in the PB2 (V203I), PB1 (R334K), nucleoprotein (G484S), and NA (V191I) genes (Table 2; Figure 1, panel B). This finding suggests recent common ancestry of the HL18NL11 variants identified in the 2 positive bats and was consistent with their detection in the same site 5 days apart. Comparison of the full coding sequence of the novel HL18NL11 variants revealed high sequence identity between the Peru and the Brazil strains, 93.5%–96.9% nucleotide identity across all 8 genomic segments (Table 2). The genomic relatedness of Peru and Brazil HL18NL11 strains was surprising given a time span of 2 years, a geographic distance exceeding 2,000 km, and 2 different bat species that tested positive in our study and the previous study (5).
All critical amino acid residues associated with influenza A virus replication and entry (4,5) were conserved between the Brazil and the Peru HL18NL11 strains, including the HA monobasic cleavage site motif PIKETR/GLF (5). Thermodynamic modeling revealed that the amino acid exchanges observed between the Brazil and Peru HL18NL11 strains did not alter the tridimensional structure of the HL and NL proteins, and neither mapped to the putative receptor binding site of the HL protein (Figure 1, panel C), nor to the putative active site of the NL protein (Figure 1, panel D) (6). This result suggests preservation of the biologic activity of these glycoproteins in different bat species and supported a broadly conserved cellular receptor of bat influenza A viruses that differs from sialic acid receptors used by avian-associated influenza A viruses (10). Significantly fewer amino acid exchanges were observed between the HL proteins of Brazil and Peru bat influenza virus than between the respective NL proteins (p = 0.007 by Fisher exact test) (Table 2). The apparently low rate of nonsynonymous substitutions in the HL-encoding genes of bat influenza A virus variants was reminiscent of strong purifying selection acting on the hemagglutinin genes in avian-specific influenza A virus strains (11). This finding may suggest comparable evolutionary dynamics between chiropteran and avian reservoirs. Definite assessments will require considerably larger datasets of bat influenza A virus strains.
Figure 2

Figure 2. Phylogenetic relationships between bat influenza A viruses from Brazil and reference viruses. Phylogenetic trees show comparison of the 8 segments of representative influenza A virus genomes (PB2, PB1, PA, HA/HL, NP,...
A. lituratus bats and A. planirostris bats, in which HL18NL11 was originally detected in Peru, represent closely related, yet genetically and morphologically clearly distinct bat species (12). The distribution of these bat species overlaps (Figure 1, panel A), potentially facilitating virus exchange across the populations. Phylogenetic analyses confirmed the close genetic relationship between Peru and Brazil HL18NL11 variants across all 8 segments (Figure 2; Appendix Table 2), suggesting lack of reassortment events according to the available data. Our data thus suggest host associations of HL18NL11 beyond the species level, comparable to genus-level host associations of other batborne RNA viruses such as coronaviruses (13).
The zoonotic potential of HL18NL11 is unclear, yet human-derived cell lines were susceptible to infection by chimeric vesicular stomatitis virus pseudotyped with HL18 (14). The abundance of A. lituratus bats within Latin America (Figure 1, panel A) may thus facilitate spillover infections into other vertebrates across an underrecognized geographic and host range. Finally, Artibeus spp. bats have been used previously for infection studies including viruses with evolutionary origins in bats, such as Middle East respiratory syndrome coronavirus (15). The relatively large body size of A. lituratus bats (≈65 g) and ease of keeping these bats under laboratory conditions may thus facilitate experimental infection studies for HL18NL11 to elucidate the exact sites of HL18NL11 replication, receptor usage, and mode of transmission.
Dr. Campos is a postdoctorate researcher affiliated with the University of Sao Paulo and Charité-Universitätsmedizin Berlin. Her research focuses on emerging viruses from bats.
Acknowledgments
We thank Mariana Cristine Pereira de Souza, Cairo Monteiro de Oliveira, and Luciano Matsumiya Thomazelli for laboratory support.
This work was supported by FAPESP (São Paulo Research Foundation) through grants 2017/20744-5, 2014/15090-8, 2014/16320-7, 2013/11006-0, and 2008/57687-0 and the European Union’s Horizon 2020 research and innovation programme through the ZIKAlliance project (grant agreement no. 734548) to J.F.D.
References
- Olsen B, Munster VJ, Wallensten A, Waldenström J, Osterhaus ADME, Fouchier RAM. Global patterns of influenza a virus in wild birds. Science. 2006;312:384–8. DOIPubMedGoogle Scholar
- Brunotte L, Beer M, Horie M, Schwemmle M. Chiropteran influenza viruses: flu from bats or a relic from the past? Curr Opin Virol. 2016;16:114–9. DOIPubMedGoogle Scholar
- Olival KJ, Hosseini PR, Zambrana-Torrelio C, Ross N, Bogich TL, Daszak P. Host and viral traits predict zoonotic spillover from mammals. Nature. 2017;546:646–50. DOIPubMedGoogle Scholar
- Tong S, Li Y, Rivailler P, Conrardy C, Castillo DA, Chen LM, et al. A distinct lineage of influenza A virus from bats. Proc Natl Acad Sci U S A. 2012;109:4269–74. DOIPubMedGoogle Scholar
- Tong S, Zhu X, Li Y, Shi M, Zhang J, Bourgeois M, et al. New world bats harbor diverse influenza A viruses. PLoS Pathog. 2013;9:e1003657. DOIPubMedGoogle Scholar
- García-Sastre A. The neuraminidase of bat influenza viruses is not a neuraminidase. Proc Natl Acad Sci U S A. 2012;109:18635–6. DOIPubMedGoogle Scholar
- Góes LGB, Campos ACA, Carvalho C, Ambar G, Queiroz LH, Cruz-Neto AP, et al. Genetic diversity of bats coronaviruses in the Atlantic Forest hotspot biome, Brazil. Infect Genet Evol. 2016;44:510–3. DOIPubMedGoogle Scholar
- Anthony SJ, Islam A, Johnson C, Navarrete-Macias I, Liang E, Jain K, et al. Non-random patterns in viral diversity. Nat Commun. 2015;6:8147. DOIPubMedGoogle Scholar
- Drexler JF, Corman VM, Wegner T, Tateno AF, Zerbinati RM, Gloza-Rausch F, et al. Amplification of emerging viruses in a bat colony. Emerg Infect Dis. 2011;17:449–56. DOIPubMedGoogle Scholar
- Zhu X, Yu W, McBride R, Li Y, Chen LM, Donis RO, et al. Hemagglutinin homologue from H17N10 bat influenza virus exhibits divergent receptor-binding and pH-dependent fusion activities. Proc Natl Acad Sci U S A. 2013;110:1458–63. DOIPubMedGoogle Scholar
- Rejmanek D, Hosseini PR, Mazet JAK, Daszak P, Goldstein T. Evolutionary dynamics and global diversity of influenza A virus. J Virol. 2015;89:10993–1001. DOIPubMedGoogle Scholar
- Larsen PA, Marchán-Rivadeneira MR, Baker RW. Speciation dynamics of the fruit-eating bats (genus Artibeus): with evidence of ecological divergence in Central American populations. In: Adams RA, Pedersen SC, editors. Bat evolution, ecology, and conservation. New York: Springer Science + Business Media; 2013. p. 315–339.
- Drexler JF, Corman VM, Drosten C. Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antiviral Res. 2014;101:45–56. DOIPubMedGoogle Scholar
- Moreira ÉA, Locher S, Kolesnikova L, Bolte H, Aydillo T, García-Sastre A, et al. Synthetically derived bat influenza A-like viruses reveal a cell type- but not species-specific tropism. Proc Natl Acad Sci U S A. 2016;113:12797–802. DOIPubMedGoogle Scholar
- Munster VJ, Adney DR, van Doremalen N, Brown VR, Miazgowicz KL, Milne-Price S, et al. Replication and shedding of MERS-CoV in Jamaican fruit bats (Artibeus jamaicensis). Sci Rep. 2016;6:21878. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleOriginal Publication Date: January 07, 2019
1These senior authors contributed equally to this article.
Table of Contents – Volume 25, Number 2—February 2019
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Jan Felix Drexler, Helmut-Ruska-Haus, Institute of Virology, Campus Charité Mitte, Charitéplatz 1, 10098 Berlin, Germany
Top