Volume 26, Number 12—December 2020
Dispatch
Circulation of 2 Barmah Forest Virus Lineages in Military Training Areas, Australia
Cite This Article
Citation for Media
Abstract
During 2017–2018, Barmah Forest virus was recovered from mosquitoes trapped in military training areas in Australia and from a soldier infected at 1 of these areas. Phylogenies of the nucleotide sequences of the envelope glycoprotein gene E2 and the 3′ untranslated region suggest that 2 lineages are circulating in eastern Australia.
With »15,000 laboratory-confirmed cases over the last decade, Barmah Forest virus (BFV) is the second most common cause of human arboviral disease in Australia, after Ross River virus (RRV) (1). BFV is a positive-sense, single-strand, enveloped RNA virus of genus Alphavirus, family Togaviridae. Other viruses in this genus include chikungunya virus, RRV, Sindbis virus, and Eastern and Western equine encephalitis viruses. BFV was first isolated in 1974 from Culex annulirostris mosquitoes trapped near Barmah Forest, northern Victoria, Australia (2); the first case of a clinical BFV infection in humans was reported in 1986 (3). Since then, BFV has been reported throughout mainland Australia and Papua New Guinea (4,5). Clinical signs and symptoms of BFV infection, including polyarthritis, arthralgia, and myalgia, are similar to but milder than those of RRV infection (6–7). Through phylogenetic analyses of the nucleotide sequences of complete E2 envelope protein genes and of the 3′ untranslated region (3′ UTR), we identified 3 BFV lineages. However, we found only 1 example of 2 of the lineages (5,8).
RRV caused epidemic polyarthritis outbreaks in military personnel in Australia during and after short military exercises in the Shoalwater Bay Training Area in northeastern Australia in 2016 and 2017 (9). The soldier in this study was among personnel who sought treatment during the 2017 outbreak with a suspected RRV infection. Signs and symptoms included rash on the face and body, nausea, headache, fatigue, lethargy, and joint and muscle pain. This retrospective study was approved by the Australian Department of Defence and Department of Veterans’ Affairs Human Research Ethics Committee (DDVA HREC), Joint Health Command Low-Risk Ethical Review Panel (no. 16-021). We obtained formal written consent from the soldier.
During a retrospective investigation of the outbreak, using PanBio ELISA kits (Abbott, https://www.abbott.com), we detected BFV IgG and IgM, but not RRV IgG and IgM, in convalescent serum samples collected 23, 28, and 38 days after onset of symptoms in the patient. After inoculating 100 µL of the serum into cultures of C6/36 mosquito cells and 2 subsequent passages in this cell line, we did not detect infectious virus in the acute-phase serum sample collected on the day of symptom onset. However, we detected BFV RNA, but not RRV RNA, using a quantitative reverse-transcription PCR assay of RNA extracted from 140 µL of the acute-phase sample using a QiaAMP Viral RNA Mini Kit (QIAGEN, https://www.qiagen.com). BFV E2 RNA was present at 4.2 ×106 copies/mL, with a forward primer 8985F (5′-AGTGTGGCAGTACAACTCCCAAT-3′) corresponding to genome position 8985–9006 and a reverse primer (5′-AAGGCACATGGATCTTTCCTTTC-3′) corresponding to genome position 9036–9058.
For sequencing, we amplified the E2 and 3′ UTR genes by reverse transcription PCR using primers E2 forward 8205F 5′-GCTGTCTGACCACTACTACCA-3′ and E2 reverse 9833R 5′-GACTTAATCACTACTAAAGATAGCG-3′, and 3′ UTR forward 10923F 5′-TCCATCCATCTCTACTACCG-3′ and reverse poly-T 5′-TTTTTTTTTTTTTTTTTTTTG-3′ designed from the nucleotide sequence of the prototype BFV strain (BH2193) and synthesized by Sigma (https://www.sigmaaldrich.com). The PCR amplifications were performed using pfu DNA polymerase (Promega, https://www.promega.com), which has 3′–5′ exonuclease proofreading activity. The PCR amplicons were separated by agarose gel electrophoresis and recovered using a rapid gel extraction system (QIAGEN). Sequencing was performed at the Australian Genome Research Facility as described elsewhere (10). Sequences were assembled and edited using Geneious version 11.2 (https://www.geneious.com). The human isolate was named SWBTA40/2017. Two strains of BFV (TullyA/2017 and WBTA/2018) isolated from mosquitoes collected in the Tully and Wide Bay military training areas in Queensland, eastern Australia, in 2017 and 2018 (Table 1), as well as 7 BFV strains collected previously in Australia during 1974–2015, were also sequenced in the same manner. All sequences were submitted to GenBank (accession no. MK169381-6 and MH618666). Seven BFV E2 gene sequences within GenBank, including that of a recent isolate from Papua New Guinea (5), were included in phylogenetic analyses.
The SWBTA40/2017 E2 comprised 1263 nt corresponding to nucleotides 8290–9552 of the genomic RNA of the BFV prototype strain BH2193. The sequence similarity among all 17 E2 sequences we examined was remarkably high, with an overall divergence of <4.1%. The nucleotide sequence of SWBTA40/2017 was most closely related to that of the BFV prototype strain BH2193, but differed from it at 4 sites, 8692 (A®G), 8835 (U®C), 9108 (A®G), and 9427 (U®C), resulting in nucleotide divergence of 0.32% (Table 2). The A®G substitution at 8692 resulted in an amino acid substitution of Asn®Asp and the U®C substitution at 9427 resulted in a Ser®Pro amino acid change. SWBTA40/2017 E2 gene diverged from TullyA/2017 in 24 (1.9%) nt positions and from WBTA/2018 in 25 (1.98%) nt positions (Table 2). The A®G substitution at 9197 resulted in a Ser®Asn change and a U®C substitution at 9433 resulted in a Phe-to-Leu change in both the TullyA and WBTA BFV strains.
Figure 1

Figure 1. Nucleotide alignment of 3′ untranslated region sequences of Barmah Forest virus strains from Australia with that of the prototype BH2193 strain using muscle alignment method in Geneious version 11.2 (...
The SWBTA40/2017 3′ UTR comprised 443 nt corresponding to nucleotides 11047–11488 of the prototype strain BH2193 but with a single-nucleotide insertion at position 11128 (Figure 1); it differed from that of the TullyA/2017 strain by 110 nt and the WBTA/2018 strain by 114 nt (Figure 1), with multiple insertions/deletions (indels) occurring in the recent TullyA/2017 and WBTA/2018 strains. The noticeable differences were the finding of 2 large indels at nucleotide positions 3–27 and 205–263 of the 3′ UTR (Figure 1). The potential influence of these indels on BFV replication and transmission warrants further study because the 3′ UTR region plays critical roles in alphaviral gene expression, replication, protein translation, and host tropism (14).
Figure 2
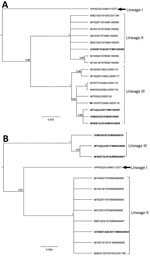
Figure 2. Phylogenies based on 17 complete BFV E2 sequences (1,263 bp) (A) and twelve 3′ untranslated region sequences (B) classify Barmah Forest virus isolates into 3 distinct lineages. We used Bayesian...
We derived phylogenies using Geneious version 11.2 with neighbor joining, maximum-likelihood, and Bayesian analysis for 17 complete E2 and twelve 3′ UTR sequences. E2 sequences segregated into 3 lineages with strong bootstrap support (Figure 2, panel A), instead of the 1 (8) or 2 (5) lineages reported previously. The lineages are numbered chronologically; the strain from the central province of Papua New Guinea from 2014, which appears to be the oldest, was denoted as lineage I (5). The isolates described in this study (SWBTA40/2017, TullyA/2017, and WBTA/2018) were placed into 2 distinct lineages, lineages II and III. The conclusion about 3 lineages was supported by the analysis of the 3′ UTR regions (Figure 2, panel B).
All amino acid substitutions were located in the C domain of the E2 protein in areas that are involved in interaction with other proteins (E1, capsid, and 6k), as well as in the process of budding of alphavirus envelope proteins from host cell membranes (11,12). BM/C583/1978 in lineage II was isolated from Culicoides midges rather than mosquitoes and may reflect BFV in a blood meal rather than this insect being a vector for BFV. Nonetheless, given the position of BFV in the phylogeny of alphaviruses, exploration of vectors other than mosquitos for this virus might be warranted.
Given the relatively recent detection of BFV in western Australia (13,14) and the basal position in phylogenetic trees of the only isolate of BFV from Papua New Guinea, the cocirculation of 2 lineages of BFV in eastern Australia points to a poor understanding of population dynamics and evolution in this virus. The ongoing replacement of strains of RRV and the appearance of strains with epidemic potential (15) suggests that BFV may warrant more detailed virological surveillance.
Dr. Liu is head of the Arbovirology Department at the Australian Defence Force Malaria and Infectious Disease Institute. His primary research interests are epidemiology, and the evolution and transmission of emerging arboviral diseases.
Acknowledgments
The authors would like to thank the soldier for participating in the study and Dennis Shanks for proofreading the manuscript.
Joint Health Command of Australian Defence Force funded this investigation. The funder had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- Australian Government Department of Health National Notifiable Diseases Surveillance System. Number of notifications of Ross River virus and Barmah Forest virus infection, Australia, in the period of 1991 to 2018 and year-to-date notifications for 2019 [cited on 2019 Oct 15]. http://www9.health.gov.au/cda/source/rpt_3.cfm
- Dalgarno L, Short NJ, Hardy CM, Bell JR, Strauss JH, Marshall ID. Characterization of Barmah forest virus: an alphavirus with some unusual properties. Virology. 1984;133:416–26. DOIPubMedGoogle Scholar
- Vale TG, Carter IW, McPhie KA, James GS, Cloonan MJ. Human arbovirus infections along the south coast of New South Wales. Aust J Exp Biol Med Sci. 1986;64:307–9. DOIPubMedGoogle Scholar
- Australian Government Department of Health National Notifiable Diseases Surveillance System. Number of notifications of Barmah Forest virus infection, Australia, in the period of 1991 to 2018 and year-to-date notifications for 2019 [cited 2019 Oct 15]. http:// http://www9.health.gov.au/cda/source/rpt_3_sel.cfm
- Caly L, Horwood PF, Vijaykrishna D, Lynch S, Greenhill AR, Pomat W, et al. Divergent Barmah Forest Virus from Papua New Guinea. Emerg Infect Dis. 2019;25:2266–9. DOIPubMedGoogle Scholar
- Jacups SP, Whelan PI, Currie BJ. Ross River virus and Barmah Forest virus infections: a review of history, ecology, and predictive models, with implications for tropical northern Australia. Vector Borne Zoonotic Dis. 2008;8:283–97. DOIPubMedGoogle Scholar
- Jeffery JAL, Kay BH, Ryan PA. Role of Verrallina funerea (Diptera: Culicidae) in transmission of Barmah Forest virus and Ross River virus in coastal areas of eastern Australia. J Med Entomol. 2006;43:1239–47. DOIPubMedGoogle Scholar
- Poidinger M, Roy S, Hall RA, Turley PJ, Scherret JH, Lindsay MD, et al. Genetic stability among temporally and geographically diverse isolates of Barmah Forest virus. Am J Trop Med Hyg. 1997;57:230–4. DOIPubMedGoogle Scholar
- Liu W, Kizu JR, Le Grand LR, Moller CG, Carthew TL, Mitchell IR, et al. Localized outbreaks of epidemic polyarthritis among military personnel caused by different sublineages of Ross River virus, northeastern Australia, 2016–2017. Emerg Infect Dis. 2019;25:1793–801. DOIPubMedGoogle Scholar
- Liu WJ, Rourke MF, Holmes EC, Aaskov JG. Persistence of multiple genetic lineages within intrahost populations of Ross River virus. J Virol. 2011;85:5674–8. DOIPubMedGoogle Scholar
- Jose J, Snyder JE, Kuhn RJ. A structural and functional perspective of alphavirus replication and assembly. Future Microbiol. 2009;4:837–56. DOIPubMedGoogle Scholar
- Li L, Jose J, Xiang Y, Kuhn RJ, Rossmann MG. Structural changes of envelope proteins during alphavirus fusion. Nature. 2010;468:705–8. DOIPubMedGoogle Scholar
- Ehlkes L, Eastwood K, Webb C, Durrheim D. Surveillance should be strengthened to improve epidemiological understandings of mosquito-borne Barmah Forest virus infection. Western Pac Surveill Response J. 2012;3:63–8. DOIPubMedGoogle Scholar
- Gyawali N, Taylor-Robinson AW, Bradbury RS, Potter A, Aaskov JG. Infection of western gray kangaroos (Macropus fuliginosus) with Australian arboviruses associated with human infection. Vector Borne Zoonotic Dis. 2020;20:33–9. DOIPubMedGoogle Scholar
- Aaskov J, Jones A, Choi W, Lowry K, Stewart E. Lineage replacement accompanying duplication and rapid fixation of an RNA element in the nsP3 gene in a species of alphavirus. Virology. 2011;410:353–9. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleOriginal Publication Date: November 08, 2020
Table of Contents – Volume 26, Number 12—December 2020
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Wen Jun Liu, Australian Defence Force Malaria and Infectious Disease Institute, Weary Dunlop Drive, Gallipoli Barracks, Enoggera, Brisbane, QLD 4051, Australia
Top