Volume 26, Number 4—April 2020
Dispatch
Person-to-Person Transmission of Andes Virus in Hantavirus Pulmonary Syndrome, Argentina, 2014
Cite This Article
Citation for Media
Abstract
Andes virus is unique among hantaviruses because it can be transmitted from person to person. This mechanism was previously supported by epidemiologic data and genetic evidence based only on partial sequences. We used full-length virus sequencing to confirm person-to-person transmission of this virus in a cluster of 3 cases in Argentina in 2014.
Pathogenic hantaviruses are members of the family Hantaviridae and genus Orthohantavirus. These viruses are responsible for hantavirus pulmonary syndrome (HPS) in the Americas. In Argentina, HPS was first described in 1995 during an outbreak in the Andean region of Patagonia, leading to the characterization of Andes virus (ANDV) (1). Since then, >1,200 cases have been confirmed in Argentina (2,3).
Hantaviruses are enveloped, single-stranded, RNA viruses with tripartite negative sense genomes. The small (S, 1.8–2.1 kb) segment encodes a nucleocapsid protein, the medium (M, 3.6–3.8 kb) a glycoprotein precursor, and the large (L, 6.5–6.7 kb) an RNA-dependent RNA polymerase (4).
Humans usually become infected with hantaviruses through inhalation of aerosolized excreta produced by infected rodents. ANDV is the unique hantavirus capable of being transmitted from person-to-person (5–7). Infection by this route takes place during the early prodromal phase, and the incubation period ranges from 9 to 40 days (8).
In previous outbreaks, genetic analysis was performed on partial sequences of ANDV, which represented ≈10% of the genome. Thus, the aim of this study was to analyze a cluster of 3 case-patients for whom epidemiologic data were available and compare complete viral genome sequences to assess person-to-person transmission or co-exposure to the same rodent population.
The occurrence of 3 clustered cases during 2014 led us to suspect person-to-person transmission. The cases were reported during a 43-day period in El Bolsón, Rio Negro. The 3 case-patients had severe disease; 2 of these case-patients (P1 and P2) died (Table 1). P1 and P2, who were twin brothers, had symptoms develop 2 weeks apart, and each sought care at a primary healthcare center (Hospital Area El Bolsón, Rio Negro). P3 was a nurse who attended P2 during this initial hospitalization. After the beginning of the cardiopulmonary phase, each patient was transferred to a high-complexity hospital in Bariloche (Hospital Zonal Bariloche). P1 died 5 days and P2 7 days after symptom onset; P3 survived.
For our investigation, we included 2 unrelated HPS case-patients (NCR1 and NCR2) from the same area for comparison. We confirmed HPS in all 5 patients by using laboratory detection of ANDV-specific IgM and reverse transcription quantitative PCR, as described (3). All patients, except P2, had ANDV-specific IgG.
For whole-genome sequencing, we extracted RNA from peripheral blood of patients and prepared sequencing libraries by using target-enrichment technology and ANDV-specific probes. As expected, we identified the South variant of ANDV in the 5 case-patients. Comparative analysis showed 100% nucleotide identity in the whole genome between the samples from patients P2 and P3 (P2/P3 genome). Patient P1 had 100% nucleotide identity in the complete S and M segments with P2/P3 but had 2 nt changes in the L segment (99.95% nucleotide identity) (Table 2); both differences were silent mutations.
On the basis of accurate epidemiologic findings, the only source of infection for P3 was her contact with P2 at Hospital Area El Bolsón. Despite the nucleotide differences between their isolates, co-exposure of P1 and P2 should not be discarded as a source of infection because these persons lived in the same house where they shared the same room and bed. However, even if one considers that these nucleotide changes were 2 silent mutations in the whole viral genome, person-to-person transmission is still the most probable way of infection for P2. A previous study reported a high degree of sequence diversity for the L segment of Puumala virus (9), which is consistent with our results.
Figure 1
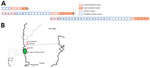
Figure 1. Temporal and geographic location of 2 cases of hantavirus pulmonary syndrome, southwestern Argentina, 2014. A) Timeline showing contact events, incubation periods, and period of illness for the 3 cases. Asterisks (*)...
Figure 2

Figure 2. Phylogenetic analysis of hantaviruses based on complete genome of Andes virus (ANDV) isolated from case-patients in Argentina, 2014, and other orthohantaviruses characterized previously. A) Small (S) segment; B) medium (M) segment;...
For further comparison, we obtained the complete sequences of 4 ANDV genomes circulating during a short period in the same area where this virus was first described (1). Nucleotide identity among strains from Argentina for the S and M segments ranged from 94.5% to 98.7%; for the L segment, it ranged from 93.6% to 98.7%. These comparisons included reference sequences for a strain from Chile. We observed a higher genetic identity between virus strains from patients P1, P2, P3, and NRC2. However, the site from which virus from NRC1 was isolated was closer to the sites of isolation of viruses from P1, P2, and P3 than to the site of isolation for virus from NRC2 (Figure 1). This finding confirmed the presence of different subtypes of the ANDV South variant co-circulating in nearby areas. Phylogenetic analysis showed that viruses from case-patients P1, P2, and P3 clustered together; NRC2 had the highest identity values for the 3 genomic segments (Figure 2).
The open reading frames encoding the nucleoprotein, glycoprotein precursor, and RNA polymerase had the same size as sizes of published sequences of ANDV (10–12). The highest degree of identity for the 4 proteins was for virus from NRC2. We compared predicted amino acid sequences with all available complete sequences of ANDV variants circulating in Argentina (South, Lech, BsAs, and Orán). Virus from P1, P2, P3, and NRC2 had 2 identical amino acid differences (T641I and T938A) in the predicted glycoprotein precursor. These differences were not found in any other sequences analyzed. Minor amino acid changes could have major effects on virus properties. Whether an amino acid substitution in viruses from HPS case-patients could determine the person-to-person transmission mechanism should be addressed by comparative analysis of higher numbers of complete virus sequences and specific studies on ANDV transmissibility.
Future studies are needed to obtain additional complete viral sequences from rodent populations co-circulating in the same geographic area. This information will enable definitive differentiation of person-to-person transmission from co-exposures in patients with similar activities of risk in disease-endemic regions, which was the case for P1 and P2, both of whom reported collecting firewood in a forested area.
We characterized the complete genome of an ANDV strain involved in a person-to-person transmission chain by using target-specific whole-genome sequencing. Our study contributed useful data for clarifying properties involved in the unusual transmissibility of ANDV. These data are crucial for optimal management of HPS case-patients and control of future outbreaks of this lethal disease.
Mr. Alonso is a virologist and a doctoral candidate at the Instituto Nacional de Enfermedades Infecciosas, Buenos Aires, Argentina. His primary research interest is hantaviruses.
Acknowledgment
We thank the physicians and epidemiologists for collecting samples and providing epidemiologic data.
References
- López N, Padula P, Rossi C, Lázaro ME, Franze-Fernández MT. Genetic identification of a new hantavirus causing severe pulmonary syndrome in Argentina. Virology. 1996;220:223–6. DOIPubMedGoogle Scholar
- Martinez VP, Bellomo CM, Cacace ML, Suarez P, Bogni L, Padula PJ. Hantavirus pulmonary syndrome in Argentina, 1995-2008. Emerg Infect Dis. 2010;16:1853–60. DOIPubMedGoogle Scholar
- Alonso DO, Iglesias A, Coelho R, Periolo N, Bruno A, Córdoba MT, et al. Epidemiological description, case-fatality rate, and trends of Hantavirus Pulmonary Syndrome: 9 years of surveillance in Argentina. J Med Virol. 2019;91:1173–81. DOIPubMedGoogle Scholar
- Hepojoki J, Strandin T, Lankinen H, Vaheri A. Hantavirus structure—molecular interactions behind the scene. J Gen Virol. 2012;93:1631–44. DOIPubMedGoogle Scholar
- Padula PJ, Edelstein A, Miguel SD, López NM, Rossi CM, Rabinovich RD. Hantavirus pulmonary syndrome outbreak in Argentina: molecular evidence for person-to-person transmission of Andes virus. Virology. 1998;241:323–30. DOIPubMedGoogle Scholar
- Martinez VP, Bellomo C, San Juan J, Pinna D, Forlenza R, Elder M, et al. Person-to-person transmission of Andes virus. Emerg Infect Dis. 2005;11:1848–53. DOIPubMedGoogle Scholar
- Ferres M, Vial P, Marco C, Yanez L, Godoy P, Castillo C, et al.; Andes Virus Household Contacts Study Group. Prospective evaluation of household contacts of persons with hantavirus cardiopulmonary syndrome in chile. J Infect Dis. 2007;195:1563–71. DOIPubMedGoogle Scholar
- Martinez VP. A major outbreak of hantavirus pulmonary syndrome caused by person-to-person transmission of Andes virus Epuyén, southwestern Argentina. Presented at: 11th International Conference on Hantaviruses; 2019 Sep 3; Leuven, Belgium.
- Razzauti M, Plyusnina A, Henttonen H, Plyusnin A. Microevolution of Puumala hantavirus during a complete population cycle of its host, the bank vole (Myodes glareolus). PLoS One. 2013;8:
e64447 . DOIPubMedGoogle Scholar - Padula PJ, Sanchez AJ, Edelstein A, Nichol ST. Complete nucleotide sequence of the M RNA segment of Andes virus and analysis of the variability of the termini of the virus S, M and L RNA segments. J Gen Virol. 2002;83:2117–22. DOIPubMedGoogle Scholar
- López N, Padula P, Rossi C, Miguel S, Edelstein A, Ramírez E, et al. Genetic characterization and phylogeny of Andes virus and variants from Argentina and Chile. Virus Res. 1997;50:77–84. DOIPubMedGoogle Scholar
- Meissner JD, Rowe JE, Borucki MK, St Jeor SC. Complete nucleotide sequence of a Chilean hantavirus. Virus Res. 2002;89:131–43. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleOriginal Publication Date: March 03, 2020
Table of Contents – Volume 26, Number 4—April 2020
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Valeria P. Martinez, Av. Velez Sarsfield 563, Buenos Aires, Argentina
Top