Volume 26, Number 7—July 2020
Dispatch
Possible Bat Origin of Severe Acute Respiratory Syndrome Coronavirus 2
Figure 1
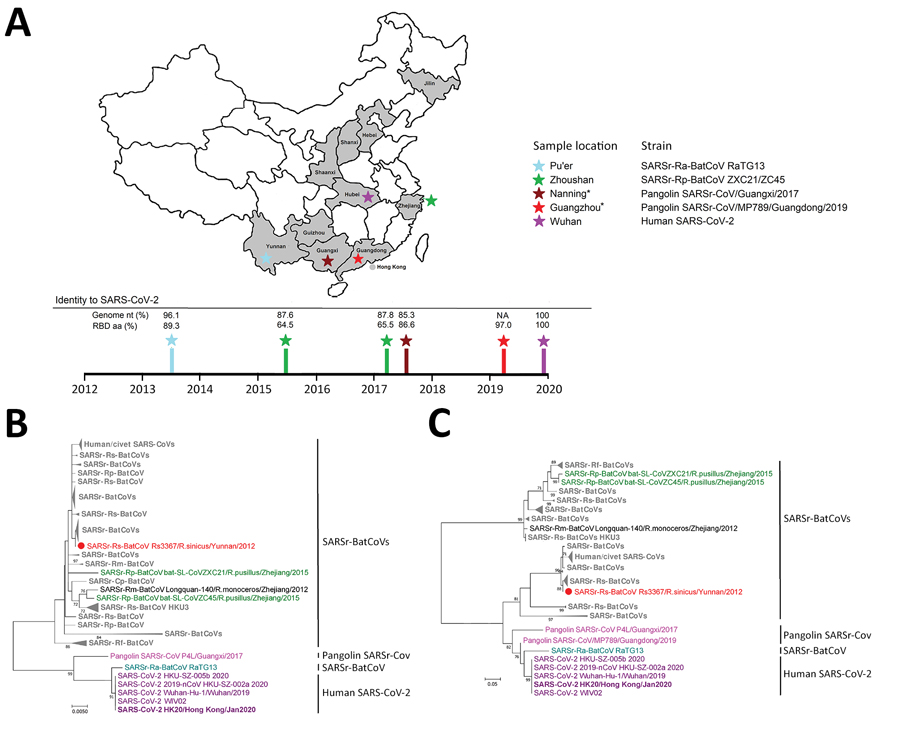
Figure 1. Geographic and phylogenetic comparisons of SARS-CoV-2 isolates with closely related viruses. A) Locations in China where SARS-CoV-2 first emerged (Wuhan), and where closely related viruses were found, including SARSr-Ra-BatCoV RaTG13 (Pu’er), Pangolin-SARSr-CoVs (Guangzhou and Nanning), and SARSr-Rp-BatCoV ZC45 (Zhoushan). Time of sampling and percentage genome identities to SARS-CoV-2 are shown. *Guangzhou and Nanning. The geographic origin of smuggled pangolins remains unknown. B, C) Phylogenetic analyses of RdRp (B) and RBD (C) domains of SARSr-CoVs. Trees were constructed by using maximum-likelihood methods with Jones-Taylor-Thornton plus gamma plus invariant sites (RdRp) and Whelan and Goldman plus gamma (RBD) substitution models. A total of 745 aa residues for RdRp and 177 aa residues for RBD were included in the analyses. Numbers at nodes represent bootstrap values, which were calculated from 1,000 trees. Only bootstrap values >70% are shown. Purple indicates SARS-CoV-2 (strain HK20 in bold); teal indicates SARSr-Ra-BatCoV RaTG13; pink indicates pangolin SARSr-CoVs; green indicates SARSr-Rp-BatCoVs ZXC21 and ZC45; red indicates SARSr-Rs-BatCoV Rs3367; black indicates SARSr-Rs-BatCoV Longquan-140; gray indicates remaining SARSr-BatCoVs. Dots indicate SARSr-BatCoVs reported to use angiotensin-converting enzyme 2 as receptor. Scale bars indicate estimated number of amino acid substitutions per 200 aa residues for RdRp and per 20 aa residues for RBD. SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; SARSr-CoV, severe acute respiratory syndrome–related coronavirus; NA, not available; RBD, receptor-binding domain; RdRp, RNA-dependent RNA polymerase.
1These authors contributed equally to this article.