Volume 27, Number 2—February 2021
Dispatch
SARS-CoV-2 Transmission between Mink (Neovison vison) and Humans, Denmark
Cite This Article
Citation for Media
Abstract
Severe acute respiratory syndrome coronavirus 2 has caused a pandemic in humans. Farmed mink (Neovison vison) are also susceptible. In Denmark, this virus has spread rapidly among farmed mink, resulting in some respiratory disease. Full-length virus genome sequencing revealed novel virus variants in mink. These variants subsequently appeared within the local human community.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has caused the ongoing coronavirus disease (COVID-19) pandemic (1). Ferrets, cats, dogs, Syrian hamsters, and nonhuman primates can be infected with the virus and, in some cases, transmit it (2); however, other species, such as pigs and chickens, appear resistant (3,4). Thus, the virus has a restricted host range. Infection with SARS-CoV-2 has occurred in farmed mink in the Netherlands (5).
In Denmark, there are »1,200 mink farms (6). Because of contacts between persons with COVID-19 and mink farms, investigation of SARS-CoV-2 infection within mink in Denmark was undertaken. We documented 3 premises in the Northern Jutland region of Denmark with SARS-CoV-2–infected mink and analyzed virus transmission in mink and the local human community.
We collected blood and throat, nasal, and fecal swab samples from mink adults and kits (Table 1); we also sampled feed and air. We assayed viral RNA by quantitative reverse transcription PCR (qRT-PCR) (7). We performed SARS-CoV-2 Ab ELISA (Beijing Wantai Biological Pharmacy Enterprise, http://www.ystwt.cn) as described (R. Lassaunière et al., unpub. data, https://doi.org/10.1101/2020.04.09.20056325). SARS-CoV-2–positive RNA samples were sequenced and sequences aligned using Mafft (https://mafft.cbrc.jp/alignment/server/index.html). Phylogenetic analysis was performed in MEGA 10.1.7 (8) using the maximum-likelihood general time reversible plus invariant sites plus gamma (2 categories) method (9).
We selected mink farms for investigation because of COVID-19 in persons linked to them. During initial visits, we sampled 30 apparently healthy adult mink; we tested adults and kits in follow-up visits. We analyzed serum samples for SARS-CoV-2 antibodies and assayed swab samples for SARS-CoV-2 RNA (Table 1; Appendix). At initial sampling, seroprevalence was high on farm 1 (>95%) and farm 3 (66%) but, in contrast, only 3% on farm 2. However, after the infection spread widely on farm 2, indicated by the increased prevalence of viral RNA (Table 1), a large increase in seroprevalence occurred, to >95%.
Air samples from farm 1 tested negative. However, on farms 2 and 3, multiple samples collected from exhaled air from mink or within 1 m of the cages scored positive, albeit with fairly high (>31) Ct values. None of the air samples collected outside the houses were positive. Feed samples collected at each farm tested negative.
Figure
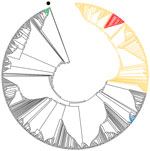
Figure. Phylogenetic tree showing relationships between genome sequences of severe acute respiratory syndrome coronavirus 2 from mink and humans at 3 mink farms in Denmark, June–July 2020 (red), and selected global...
We also sequenced SARS-CoV-2 RNA from samples from each mink farm. The viruses found on farms 1–3 were very similar (Table 2). These sequences and those from humans (H1–H9) linked to the infected farms grouped within the European 20B clade of the global SARS-CoV-2 tree (10,11) (Figure; Appendix Table 1). We deposited the SARS-CoV-2 genome sequences of virus from farm 1 (SARS-CoV-2/mink/DK/AD3_Farm1/2020) in GenBank (accession nos. MT919525–36). The sequences closely matched those of a human case, diagnosed in mid-May, with a direct epidemiologic link to farm 1. This index sequence (only 91% complete) matched the mink viruses at nt 15656 (rare globally) but had A at nt 22920 (Table 2). The nt 25936 in the index case could not be determined. The local phylogeny (Appendix Figure) showed that mink sequences from farm 1 fell into 3 subclusters (defined by the nucleotide changes at positions 5421 and 22920), but sequences from linked humans (H1–H9) and mink in farms 2 and 3 were within subcluster 2 (Appendix Figure).
We found 9 to 11 nt differences (mainly nonsynonymous) between the mink sequences in Denmark and the Wuhan-Hu-1 reference sequence (Table 2). One mutation at nt 23403 (resulting in substitution D614G in the spike protein) was present in all sequences from mink in Denmark and the Netherlands, except for NB02 from the Netherlands (Table 2) and was predominant in the human population in Jutland (Appendix Table 1) and globally (12). However, another mutation (nt C25936T [as cDNA] encoding H182 to Y within ORF3a) appeared in all mink sequences from Denmark (Table 2) and in human cases (H1–H9) linked to them. This change was not found in human SARS-CoV-2 sequences from Jutland before June 10, 2020 (Appendix Table 1), but reached »40% frequency during June 10–July 1, 2020 (Table 2; Appendix Table 2). This mutation has been found only rarely in other SARS-CoV-2 sequences (11) (Appendix Table 1) but was in mink farm NB03 from the Netherlands (SARS-CoV-2/mink/NED/NB03_index/2020; GenBank accession no. MT457400.1).
another mutation in the spike gene (A22920T, encoding Y453 to F) was present in 4 of 8 sequences from farm 1, in all sequences from farms 2 and 3, and in 5 of 6 sequences from farm NB02 in the Netherlands (5). This change was not in the index case or the human population anywhere before June 10 but was subsequently detected in farm-linked humans (H1–H9) and in Jutland (Table 2; Appendix Table 2). Finally, the mutation in the open reading frame 1b gene (C15656T, encoding T730 to I) was present only in mink/human sequences from Denmark (Table 2) and a sequence from New Zealand (Appendix Table 1).
A high proportion of mink on farms can be infected with SARS-CoV-2 within a few days, which may provide major virus exposure to persons working with mink. The infections we describe here occurred with little clinical disease or increase in death (Appendix), making it difficult to detect the spread of infection; thus, mink farms could represent a serious, unrecognized animal reservoir for SARS-CoV-2. There is no evidence for spread of the virus outside of farm buildings, either in Denmark or in the Netherlands (5), except by infected persons. However, there appears to be some risk of virus transmission to persons working with infected mink as well as for their contacts and thus, indirectly, for the public.
On farm 1, the virus had probably been introduced some weeks before detection (Table 1). On farm 2, the low frequency (4%) of seropositivity and the high proportion of qRT-PCR positive animals at second sampling (Table 1) suggested that the virus had been recently introduced but was spreading. Indeed, a third sampling (8 days later) showed a much higher seroprevalence (>90%). Conceivably, the variant viruses that appeared in farm 1 and spread to farms 2 and 3 may be better adapted to mink and thus able to transmit rapidly. The infection at farm 3 was detected relatively late, with a high seroprevalence (66%) at first visit.
A likely scenario for the spread of infection in mink in Denmark is that the index human case-patient who had nt T15656 introduced it into farm 1. Initially, we observed sequence heterogeneity at nt 22920 in mink on farm 1, but subsequently, we detected only the variant form (T22920) on farms 2 and 3 and in subsequent linked human cases (H1–H9) (Table 2). Remarkably, this heterogeneity also occurred on farm NB02 in the Netherlands. This change, possibly together with the mutation at nt 25936 (Table 2), may represent virus adaptation. It is not yet established whether these changes confer advantages in mink, but the variant viruses in farm 2 spread rapidly. It seems that the variant viruses on farm 1 spread to >1 human and were then transmitted, presumably by human–human contact, to other persons and to farms 2 and 3. The change at nt 22920 results in substitution Y453F in the S-protein (Table 2). This Y-residue, within the receptor-binding motif of the S-protein, is highly conserved among SARS-related coronaviruses and is close to residue L455 that is critical for interaction with the cellular ACE2 receptor (13).
Dr. Hammer, an associate professor at the University of Copenhagen, is a veterinary pathologist with special interest and expertise in pathological methods applied in diagnostics, research, and surveillance of diseases in fur animals and wildlife. Her research focus has been mainly on viral diseases of carnivorous species.
Acknowledgment
We thank Mads Albertsen for guidance with Nanopore sequencing and Henrik B. Krarup for providing human samples containing SARS-CoV-2. We gratefully acknowledge the provision of genetic sequence data shared via GISAID (https://www.gisaid.org; see [[ANCHOR###SD1###Appendix###Anchor]] Table 3). We also thank Amalie E. Bedsted and Thea Kristensen for careful reading of the manuscript.
References
- Johns Hopkins University. Coronavirus resource center. 2020 [cited 2020 Nov 11]. https://coronavirus.jhu.edu
- Cohen J. From mice to monkeys, animals studied for coronavirus answers. Science. 2020;368:221–2. DOIGoogle Scholar
- Schlottau K, Rissmann M, Graaf A, Schön J, Sehl J, Wylezich C, et al. SARS-CoV-2 in fruit bats, ferrets, pigs, and chickens: an experimental transmission study. Lancet Microbe. 2020;1:e218–25.
- Shi J, Wen Z, Zhong G, Yang H, Wang C, Huang B, et al. Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS–coronavirus 2. Science. 2020;368:1016–20. DOIGoogle Scholar
- Oreshkova N, Molenaar RJ, Vreman S, Harders F, Oude Munnink BB, Hakze-van der Honing RW, et al. SARS-CoV-2 infection in farmed minks, the Netherlands, April and May 2020. Euro Surveill. 2020;25:
2001005 . DOIGoogle Scholar - Miljø-og Fødevareministeriet, Fødevarestyrelsen [in Danish]. 2020 [cited 2020 Nov 11]. https://chr.fvst.dk/chri/faces/frontpage?_adf.ctrl-state=31prh7gzs_3
- Corman VM, Landt O, Kaiser M, Molenkamp R, Meijer A, Chu DK, et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. 2020;25:
2000045 . DOIGoogle Scholar - Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol Biol Evol. 2018;35:1547–9. DOIGoogle Scholar
- Nei M, Kumar S. Molecular evolution and phylogenetics. Oxford (UK): Oxford University Press; 2000.
- Alm E, Broberg EK, Connor T, Hodcroft EB, Komissarov AB, Maurer-Stroh S, et al. Geographical and temporal distribution of SARS-CoV-2 clades in the WHO European Region, January to June 2020. Euro Surveill. 2020;25:
2001410 . DOIGoogle Scholar - Nextclade. 2020 [cited 2020 Nov 11]. https://clades.nextstrain.org
- Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVD-19 virus. Cell. 2020;182:812–27.e19. DOIGoogle Scholar
- Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H, et al. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;581:221–4. DOIGoogle Scholar
Figure
Tables
Cite This ArticleOriginal Publication Date: November 18, 2020
Table of Contents – Volume 27, Number 2—February 2021
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Anette Bøtner, Department of Veterinary and Animal Sciences, University of Copenhagen, Grønnegårdsvej 15, 1870 Frederiksberg C, Denmark
Top