Volume 27, Number 4—April 2021
Research
Genomic Surveillance of a Globally Circulating Distinct Group W Clonal Complex 11 Meningococcal Variant, New Zealand, 2013–2018
Cite This Article
Citation for Media
Abstract
Genomic surveillance is an essential part of effective disease control, enabling identification of emerging and expanding strains and monitoring of subsequent interventions. Whole-genome sequencing was used to analyze the genomic diversity of all Neisseria meningitidis isolates submitted to the New Zealand Meningococcal Reference Laboratory during 2013–2018. Of the 347 isolates submitted for whole-genome sequencing, we identified 68 sequence types belonging to 18 clonal complexes (CC). The predominant CC was CC41/44; next in predominance was CC11. Comparison of the 45 New Zealand group W CC11 isolates with worldwide representatives of group W CC11 isolates revealed that the original UK strain, the 2013 UK strain, and a distinctive variant (the 2015 strain) were causing invasive group W meningococcal disease in New Zealand. The 2015 strain also demonstrated increased resistance to penicillin and has been circulating in Canada and several countries in Europe, highlighting that close monitoring is needed to prevent future outbreaks around the world.
Neisseria meningitidis, a gram-negative bacterium, is the causative agent for meningococcal meningitis and septicemia and has been associated with isolated cases, outbreaks, and epidemics worldwide (1). The rapid progression of invasive meningococcal disease (IMD) and its high incidence of severe illness and death make IMD a feared and closely monitored disease. N. meningitidis is classified into 12 groups on the basis of capsular polysaccharide structure, but 6 groups (A, B, C, W, X, and Y) cause most life-threatening IMD (2). Most meningococcal disease is caused by hyperinvasive lineages belonging to specific clonal complexes (CC) as defined by multilocus sequence typing (MLST) (3), including CC32, CC41/44, CC11, and CC5 (4). Obtaining complete genetic information and resolving the evolutionary relationships of invasive pathogens are vital for identifying the origins and expansion of new pathogenic strains.
Group W CC11 (W:CC11) meningococci emerged as a global cause of IMD after an outbreak in Mecca, Saudi Arabia, in 2000 (5). High levels of group W disease have recently occurred in many countries and regions, including the United Kingdom (6), Sweden (7), Australia (8), and North America (9,10), and whole-genome sequencing (WGS) has been used to investigate its spread. Core-genome MLST found that group W:CC11 isolates were distinct from groups B and C sequence type (ST) 11 isolates (11). Currently, 4 major W:CC11 strains belonging to 2 major lineages are circulating globally: the Hajj strain sublineage, including the Hajj strain (12), and the South America strain sublineage, which includes the South America strain that emerged in 2003 in southern Brazil (13); the UK strain, a variant of the South America strain; and the 2013 UK strain (14), which has expanded into several countries (8,9,15).
During 1991–2008, New Zealand experienced a prolonged epidemic of IMD; most cases were caused by a single N. meningitidis group B strain (NZMenB), defined by PorA type P1.7–2,4 and belonging to CC41/44 (16,17). A strain-specific vaccine, MeNZB, was introduced in 2004 (18) and withdrawn in 2008 after rates of IMD decreased (19). The NZMenB strain continues to cause about one third of meningococcal infections in New Zealand, but other group B, C, W, and Y strains are also circulating (20). Recently, incidence of IMD caused by W:CC11 has increased in New Zealand. To learn more about the genetic diversity associated with IMD after the epidemic, we used WGS to analyze 347 isolates collected during 2013–2018. We determined their clonal relationship and compared the genomes of the New Zealand W:CC11 isolates with global representatives of W:CC11 lineages.
Surveillance and Epidemiologic Analysis
Meningococcal disease is a notifiable disease in New Zealand; all IMD cases are referred to the Meningococcal Reference Laboratory at the Institute of Environmental Science and Research (ESR) for routine grouping using slide agglutination or PCR (21). For this study, disease incidence and case demographics were derived from notification data extracted from the national notifiable disease surveillance database. Annual population denominators were taken from Statistics New Zealand. We used R version 3.4.4 (https://www.r-project.org) to perform all statistical tests. Shannon–Wiener diversity index was calculated by using vegan version 2.5.6 (22).
WGS
We analyzed all available meningococcal isolates from 2013–2018 in New Zealand by WGS (Appendix 1 Table 1). Genomic DNA was purified by using the Gentra Puregene Yeast/Bact. Kit (QIAGEN, https://www.qiagen.com) or High Pure PCR Template Preparation Kit (Roche, https://www.roche.com) according to the manufacturer’s protocols. A 10-µL loop of bacteria, grown overnight on Columbia blood agar plates (Fort Richard Laboratories, Auckland, NZ), was suspended in 300 µL of lysis buffer and heat-killed at 56°C for 1 h. We then quantified DNA by using the Quant-iT PicoGreen dsDNA assay kit (Thermo Fisher Scientific, https://www.thermofisher.com) and constructed libraries by using Nextera-XT DNA Library Preparation Kit (Illumina, https://www.illumina.com). Paired-end sequencing of 2 × 150 bp was performed on the Illumina platform at ESR. Read data are available for download from the National Center for Biotechnology Information Sequence Read Archive (Bioproject accession no. PRJNA592848) and from PubMLST (https://pubmlst.org).
Genomic Analysis
Raw reads were quality trimmed by using Trimmomatic version 0.32 (23) to remove adapters, low quality bases (<Q20), and reads shorter than 60 bp. SPAdes version 3.10.1 (24) was used for assembly, and contigs >200 bp were kept. Assembled contigs were used for in silico MLST, capsular grouping, and antigen typing by using meningotype version 0.82-β (25). Single-nucleotide polymorphisms (SNPs) were identified by aligning quality-trimmed paired-end reads to the N. meningitidis reference sequence FAM18 (GenBank accession no. AM421808.1) and using Bowtie2 version 2.2.5 (26). Alignments were processed with Picard-tools (27) to remove duplicated reads and assessed with Qualimap version 2.2.1 (28) (Appendix 1 Table 2). We used FreeBayes version 1.2.0–2-g29c4002 (E. Garrison, unpub. data, https://arxiv.org/abs/1207.3907) to detect sequence variations among isolates with these settings: ploidy = 1, at 20× minimum depth and 70% minimum variant allele frequency. We filtered variants by using vcflib (29) and vcftools version 0.1.12b (30) and removed sites located in the tandem repeat regions by using the intersect function from BEDTools version 2.23.0 (31). For datasets that included only assembled genomes, we used Parsnp version 1.1.2 (32) to perform core-genome alignment and FAM18 (AM421808.1) as reference.
Phylogenetic Analysis
We constructed phylogenetic analyses from core SNP alignment by using the maximum-likelihood method under the general time reversible substitution model with RAxML version 8.2.12 (33) and estimated the relative robustness of the clades with 200 bootstrap replicates (34). We used ClonalFrameML version 1.25 (35) to construct recombination-corrected phylogeny on the basis of core SNP tree topology. For datasets that included only assembled genomes, we generated a maximum-likelihood phylogenetic tree by using RAxML on the basis of the core-genome alignment with 200 bootstrap replicates. Phylogenies were annotated by using iTOL (36).
Selection of Global Group W:CC11 Isolates
We compared New Zealand group W:CC11 isolates with 153 short-read datasets and 30 draft assemblies. Short-read data in the European Nucleotide Archive were selected to represent all major lineages of group W:CC11. We followed studies by Lucidarme et al. (14) and Tsang et al. (9) and randomly chose 1–3 isolates per year per country. We selected 153 datasets including 151 group W isolates and 2 group C isolates (ERR557598 and ERR976806), which were used as an outgroup to study the genetic relationship within group W:CC11.
We used the PubMLST Neisseria database to identify other isolates closely related to the New Zealand ST11 isolates (37). As of February 18, 2019, a total of 30 isolates were within the 50 mismatch thresholds to NZ17MI0022 based on comparison of the N. meningitidis core-genome MLST (38). Draft assemblies of these 30 isolates were downloaded from PubMLST. Short-read data were available for 3 of these 30 isolates. We compiled detailed information on sequences used for phylogenetic analysis (Appendix 1 Table 3).
Antimicrobial Susceptibility Testing of New Zealand Group W:CC11 Isolates
Of the 45 New Zealand invasive group W:CC11 isolates, 42 were assessed for susceptibility to penicillin, ciprofloxacin, ceftriaxone, and rifampin. MICs were determined by gradient strip on Mueller-Hinton agar with 5% sheep blood and interpreted according to the Clinical and Laboratory Standards Institute breakpoints (39). Assembled contigs were used for in silico penA typing by using the PubMLST Neisseria database.
Epidemiology of Meningococcal Disease in New Zealand, 2013–2018
Figure 1
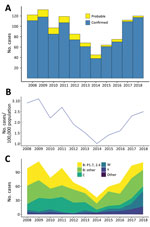
Figure 1. Epidemiology of meningococcal disease, New Zealand, 2013–2018. A) Number of confirmed and probable cases. B) Number of cases per 100,000 population of meningococcal disease. C) Proportion of meningococcal disease by...
During 2013–2018, a total of 484 cases of meningococcal disease were reported, including 456 confirmed and 28 probable cases, for a confirmation rate of 94.2% (Figure 1, panel A; Appendix 1 Table 4). Rates of IMD (Figure 1, panel B) continued to fall after the end of the group B epidemic in 2008 (coefficient = −2.619, R2 = 0.6947; p = 0.0245) but increased during 2014–2018 (coefficient = 2.481, R2 = 0.9567; p = 0.0025). Inflection point of the time series was identified by breakpoint analysis and using the R package strucchange (40) and tested by the Chow test in R by using monthly disease rates (cases/100,000) during January 2008–December 2018 (Appendix 1 Table 5). The test identified the inflection point as October 2013 (95% CI January–December 2013).
Group B meningococci continue to be responsible for most IMD cases in New Zealand. Among the 438 confirmed cases analyzed by ESR during 2013–2018 were 265 (54.7%) group B (100 NZMenB and 165 other group B) cases, 58 (13.2%) group C cases, 61 (13.9%) group W cases, and 47 (10.7%) group Y cases (Figure 1, panel C; Appendix 1 Table 6). To compare whether significant changes occurred during 2013–2018, we used a 2-proportion Z-test in R to compare the proportion of diseases caused by each group. We found that the proportion of group W disease in 2018 (33/133) was significantly greater than that in 2013 (5/58, χ2 = 8.2415; p = 0.002047). In contrast, the proportion of disease caused by group C in 2018 (10/133) is significantly less than that of 2013 (17/58, χ2 = 10.578; p = 0.00057).
Figure 2
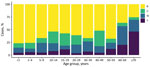
Figure 2. Age group distribution of meningococcal disease, by isolate group, New Zealand, 2013–2018
The rate of IMD continues to be highest among patients <1 year of age (from 10.2/100,000 population in 2014 to 23.1/100,000 population in 2017); rates are second highest among children 1–4 years of age (from 5.2/100,000 population in 2014 to 9.8/100,000 population in 2017). Adults >60 years of age are more affected by group W and Y disease (Figure 2; Appendix 1 Table 7).
Clonal Distribution of Circulating Meningococci
We performed WGS and in silico typing on the 347 New Zealand meningococcal isolates (288 invasive and 59 noninvasive) collected during 2013–2018. The WGS dataset includes 175 group B isolates (65 NZMenB and 110 other group B), 48 group C isolates, 2 group E isolates, 64 group W isolates, 47 group Y isolates, 1 group X isolate, and 8 nongroupable isolates. MLST analysis showed that the 347 isolates contained 49 known STs. The 10 most common STs were ST11 (85), ST154 (43), ST23 (21), ST42 (17), ST32 (16), ST213 (15), ST1655 (15), ST1572 (11), ST6058 (8), and ST22 (7). There were 39 other STs with a single isolate and 19 unassigned STs (submitted through this project). The STs identified belonged to 18 clonal complexes. A total of 93 unique strains were defined by combination of group:PorA-variable region (VR)1, PorA-VR2:FetA:CC, with a Shannon–Wiener diversity index of 3.516. Bexsero antigen sequence types analysis showed 69 unique types with 23 isolates in which type could not be determined because of the absence of required loci (Appendix 1 Table 1).
Figure 3

Figure 3. Phylogenetic analysis of New Zealand Neisseria meningitidisisolates, 2013–2018. Maximum-likelihood phylogeny was constructed by using a generalized time reversible substitution model and core single-nucleotide polymorphism alignments with RAxML version...
Maximum-likelihood phylogeny constructed from 75,187 core SNP alignments (by using N. meningitidis FAM18 as the reference genome) showed 17 highly supported clades (with >90% bootstrap value), 15 of which were consistent with the CC definition (Figure 3). One clade of group B isolates could not be assigned to a CC because its ST had not been assigned a CC designation; it is noted as N1 on the phylogenetic tree. CC11 is the predominant CC consisting of 40 group C and 54 group W isolates; the second most common was CC41/44, consisting of 90 group B and 1 nongroupable isolates.
Figure 4

Figure 4. Diversity and prevalence of PorA variable region (VR) variants in common Neisseria meningitidisstrains in New Zealand, 2013–2018. PorA VR1 and VR2 variant diversity and numbers of common strains...
Since the NZMenB epidemic, New Zealand has used a combination of PorA subtype with group information to define N. meningitidis strains. We integrated the PorA and CC information to identify the strains currently circulating in New Zealand (Figure 4).
Phylogenetic Analysis of New Zealand W:CC11 Isolates
Rates of group W disease in 2018 were higher than in 2013. Most sequenced group W isolates in 2018 (30/35) belonged to CC11. To understand how New Zealand W:CC11 isolates relate to the global group W lineages, we analyzed New Zealand W:CC11 in the context of major W:CC11 lineages. We included 196 CC11 isolates in this analysis (151 downloaded from public databases and 45 invasive isolates from New Zealand). The mapping rate was 92.4%–99.3% when N. meningitidis FAM18 (AM421808.1), a representative of CC11, was used as a reference. The mean genome depth was 152X, with 84.8%–97.7% of loci covered at >20-fold (Appendix 1 Table 2).
Figure 5

Figure 5. Phylogenetic position of New Zealand group W clonal complex 11 (W:CC11) Neisseria meningitidis isolates within the global W:CC11 major lineages. Maximum-likelihood phylogeny was generated by RAxML version 8.2.12 (...
We used core SNP (48,507 bps) alignment to construct the phylogenetic relationship of New Zealand W:CC11 isolates within the global W:CC11 (Figure 5). Phylogeny was rooted with 2 group C CC11 isolates. An unrooted neighbor-net phylogeny from the same dataset is shown in Appendix 2 Figure 1. Recombination-corrected phylogeny from the same dataset is shown in Appendix 2 Figure 2. Excluding the basal older sublineages, all other isolates formed 2 strongly supported clades. Clade I corresponds to the previously identified Hajj sublineage, and clade II corresponds to the previously identified South America sublineage. All New Zealand CC11 isolates were located within clade II: 4 clustered with the original UK strains, 12 clustered with the 2013 UK strain, and the other 29 formed a separate cluster with the 2 isolates from the United Kingdom and 1 isolate from Ireland (Figure 5).
To determine whether other isolates are closely related to the new New Zealand cluster, we searched PubMLST and found 27 additional isolates within 50 allele (the cgMLST 0.1 scheme) differences to NZ17MI0022. Because only assembled contigs were available for these isolates, we used a core-genome alignment approach to analyze their relationship within clade II of the W:CC11 isolates. All additional 27 W:CC11 isolates clustered with the New Zealand cluster with high bootstrap support (Appendix 2 Figure 3). The 27 isolates are from Canada and 6 countries in Europe (Appendix 1 Table 3).
Both phylogenetic analyses suggest that the new New Zealand cluster is part of the W:CC11 South America strain sublineage derived from the original UK strain. Because the earliest isolate of the new variant was identified in 2015, we named it the 2015 strain of the W:CC11 South America strain sublineage (the 2015 strain).
Epidemiology of the 2015 Strain
Similar to the 2013 UK strain, the 2015 strain is associated with higher death rates. During 2017–2018, the 2015 strain was associated with a death rate of 17.8% (6/34 cases) in New Zealand, higher than the rate of 5.9% (10/170 cases) for other groups in the same period (p = 0.03 by Fisher exact test). The 2015 strain also disproportionally affected older adults; 26% of total 2015-strain cases affected adults >60 years of age (9/34), whereas 5.8% (7/121) of total group B cases affected adults in that age group (p = 0.01466 by Fisher exact test).
2015 Strain and Penicillin Susceptibility
In 2016, Mowlaboccus et al. (41) described a W:CC11 variant circulating in Australia that demonstrated intermediate resistance or was resistant to penicillin and had penA allele 253. To examine whether the New Zealand 2015-strain isolates were also resistant to penicillin, we tested the antimicrobial susceptibility of 42 invasive New Zealand W:CC11 isolates in this study. All 42 isolates were susceptible to ciprofloxacin (MIC <0.03 mg/L), ceftriaxone (<0.12 mg/L), and rifampin (<0.5 mg/L) (Appendix 1 Table 8). We observed variation in penicillin susceptibility among the 42 isolates (Appendix 1 Table 8) by using Clinical and Laboratory Standards Institute breakpoints. Of the 15 isolates belonging to either the original UK strain or the 2013 UK strain, all were susceptible to penicillin (<0.06 mg/L). Of the 27 isolates belonging to the new 2015 strain, 12 displayed intermediate resistance (0.12–0.25 mg/L) and 15 were resistant (>0.5 mg/L). The 2015-strain isolates were significantly more resistant to penicillin (p<0.00001 by Fisher exact test). All 27 New Zealand 2015-strain isolates had penA allele 253, the same allele described in the Australia study (41). In the larger dataset that included 30 international 2015-strain isolates, all but 1 isolate had penA allele 253 (Appendix 1 Table 3).
We comprehensively analyzed N. meningitidis in New Zealand during 2013–2018 to describe its population structure after the NZMenB epidemic. We examined the rate of IMD and clonal distribution of circulating isolates. We also offer evidence that a distinct variant of W:CC11 is circulating globally and has been causing IMD since 2015.
Meningococcal disease continues to substantially affect the health of persons in New Zealand. In comparison with other developed countries (such as the United States, the United Kingdom, and the Netherlands, which have reported ≤1 case/100,000 population) (42–45), New Zealand still has a high rate of IMD notification. Although the rate of meningococcal disease in 2018 (2.5 cases/100,000 population) was substantially lower than the peak rate observed during the epidemic (17.4 cases/100,000 population in 2001), rates of IMD in New Zealand have increased since 2014 (Appendix 2 Figure 1).
The distribution of group Y disease and group W disease has changed over the past decade. Incidence of group Y disease has been increasing since the late 1990s in the Americas and since 2010 in Europe (46,47). The number and proportion of group Y disease in New Zealand began to increase in 2013 (2/85 [2%] in 2012 to 4/68 [6%] in 2013). The increase in group W:CC11 disease here was not significant until 2017 and 2018, when the number of group W cases more than tripled, from 10 to 33. In the United Kingdom, the original UK strain emerged in 2009; the descendant 2013 strain emerged in 2013 and expanded to other countries thereafter (15). These data suggest that IMD trends in New Zealand follow global trends with some delay, possibly because of geographic isolation. Therefore, for IMD monitoring in New Zealand, continued detailed typing of meningococcal isolates is critical for obtaining comparable data for participation in global meningococcal surveillance.
By using group and PorA type for strain definition, the diversity index for 2013–2018 period meningococci is slightly higher (Shannon–Wiener index 2.98) than that for 2008–2012, the 5-year period following the end of the epidemic (Shannon–Wiener index 2.81; data not shown). Since 2012, nonepidemic group B cases have regularly surpassed epidemic cases; 47% of group B cases were caused by nonepidemic strains during 2008–2011, and 63% were caused by nonepidemic strains during 2012–2018. The top 3 strains circulating within the nonepidemic group B cases were B:P1.22,14:F5–5:CC213 (13 isolates), B:P1.7,16–26:F3–3:CC32 (13 isolates), and B:P1.7–12,14:F1–7:CC1572 (12 isolates). All 3 strains are present in PubMLST. B:P1.22,14:CC213 is a common strain, however, only 2 isolates contain the FetA-VR:5–5 allele. PubMLST has 41 B: P1.7,16–26:F3–3:CC32 isolates and 6 B:P1.7–12,14:F1–7:CC1572 isolates (accessed November 3, 2020). Taken together, these data suggest that the meningococcal population in New Zealand has become more diverse after the group B epidemic. Comparison of New Zealand with other countries is challenging because no comprehensive public database exists that contains all IMD cases with fine-typing information. The United Kingdom, however, has been depositing most of their IMD typing information into PubMLST since July 2010, and for 2013–2017, the dataset contains 2,790 records and 649 unique strains (group:PorA-VR1,PorA-VR2:FetA:CC, Shannon–Wiener index 4.45). This information suggests that the isolates that cause IMD are less diverse in New Zealand, which may reflect its smaller and more distributed population structure.
The clonal expansion of a new penicillin-resistant clade of W:CC11 was first identified in Australia in 2016 (41). Four isolates (PubMLST identification nos. 41966, 42206, 42409, 50313) that were found to be closely related to the Australia clade (cluster B) (42) are part of the 2015-strain cluster (Appendix 2 Figure 3), suggesting that the Western Australia clone belongs in the 2015-strain cluster. In our study, the New Zealand 2015-strain isolates were significantly less susceptible to penicillin compared with New Zealand isolates belonging to the original UK or the 2013 UK strain (Appendix 1 Table 8). The penA 253 allele was hypothesized to play a role in decreasing penicillin susceptibility in the new clade of W:CC11 found in Australia (41). All isolates in the 2015 variant cluster, except ERR1994517 from the United Kingdom, have the penA 253 allele (Appendix 1 Table 3), suggesting that penicillin resistance is a common characteristic of the 2015 strain. The presence of the penA 253 allele has consequences for the choice of antimicrobial drug to treat IMD. At the end of 2018, a high rate of W:CC11 disease in the Northland area of New Zealand triggered a local vaccination campaign against the disease. Most of these cases were caused by the 2015 strain. Partly because of the increased penicillin resistance of the 2015 strain, in 2018 the New Zealand Ministry of Health recommended ceftriaxone as the first-choice antimicrobial drug for patients with suspected meningococcal disease (48).
W:CC11 meningococci are emerging as a diversifying lineage with several new strains occurring in different geographic locations (10,11,41). Phylogenetic analyses suggest that the 2015 strain is part of the major South America–UK (clade II) lineage (Figure 3; Appendix 2 Figure 3), most likely representing a clonal expansion from a single variant within the original UK strain. The increase in New Zealand group W cases is mainly attributable to the 2015 strain identified in this study. In contrast, the 2013 UK strain was largely responsible for the increases seen in Europe (15). We found the 2013 UK strain circulating in New Zealand but in low numbers (3/10 cases in 2017 and 6/33 cases in 2018). For reasons unknown, the 2015 strain, rather than the 2013 UK strain, is expanding in New Zealand. Invasive disease provides no selective advantage for meningococcal bacteria, and a strain must remain in carriage in order to expand in a population. The 2015 strain might have originated in the Australia or New Zealand region and is therefore better adapted to the population, either because of more favorable host immunity or climate and living conditions. Its carriage may have precluded expansion of the 2013 UK strain.
Genomic surveillance of N. meningitidis has revealed in great detail the genetic diversity and population structure of circulating meningococci in New Zealand; this more refined surveillance enables the tracking of specific strains that are identifiable only by high-resolution phylogenetic analysis. By using this approach, we identified a distinct globally circulating W:CC11 strain, which would not have been possible without genomic information. Our results emphasize the value of obtaining complete genetic information for invasive pathogens and resolving their global evolutionary relationship for identifying the origin and expansion of new pathogenic variants or strains. Such information will benefit surveillance and can be used to help prevent and control future epidemics.
Dr. Yang is a postdoctoral researcher at the Institute of Environmental Science and Research investigating the population genomics and evolution of the bacterial pathogen N. meningitidis. She is interested in using genome sequencing to address evolutionary questions such as tracing evolutionary history and population dynamics, as well as understanding genetic determinants of phenotypic variation.
Acknowledgments
We thank Yoryea Mantziou for her assistance in DNA sequencing; the staff of the Antibiotic Reference Laboratories, Institute of Environmental Science and Research, for antimicrobial susceptibility testing; and Kristin Dyet and Joanne Hewitt for their thoughtful comments and feedback.
This study was funded by Health Research Council grant no. 17-364, New Zealand.
This study used data from routine meningococcal surveillance activities conducted by the Institute of Environmental Science and Research under contract to the New Zealand Ministry of Health.
References
- Halperin SA, Bettinger JA, Greenwood B, Harrison LH, Jelfs J, Ladhani SN, et al. The changing and dynamic epidemiology of meningococcal disease. Vaccine. 2012;30(Suppl 2):B26–36. DOIPubMedGoogle Scholar
- Harrison LH, Trotter CL, Ramsay ME. Global epidemiology of meningococcal disease. Vaccine. 2009;27(Suppl 2):B51–63. DOIPubMedGoogle Scholar
- Maiden MCJ, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, et al. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A. 1998;95:3140–5. DOIPubMedGoogle Scholar
- Waśko I, Hryniewicz W, Skoczyńska A. Significance of meningococcal hyperinvasive clonal complexes and their influence on vaccines development. Pol J Microbiol. 2015;64:313–21. DOIPubMedGoogle Scholar
- Mustapha MM, Marsh JW, Krauland MG, Fernandez JO, de Lemos APS, Dunning Hotopp JC, et al. Genomic epidemiology of hypervirulent serogroup W, ST-11 Neisseria meningitidis. EBioMedicine. 2015;2:1447–55. DOIPubMedGoogle Scholar
- Ladhani SN, Beebeejaun K, Lucidarme J, Campbell H, Gray S, Kaczmarski E, et al. Increase in endemic Neisseria meningitidis capsular group W sequence type 11 complex associated with severe invasive disease in England and Wales. Clin Infect Dis. 2015;60:578–85. DOIPubMedGoogle Scholar
- Eriksson L, Hedberg ST, Jacobsson S, Fredlund H, Mölling P, Stenmark B. Whole-genome sequencing of emerging invasive Neisseria meningitidis serogroup W in Sweden. J Clin Microbiol. 2018;56:e01409–17. DOIPubMedGoogle Scholar
- Martin NV, Ong KS, Howden BP, Lahra MM, Lambert SB, Beard FH, et al.; Communicable Diseases Network Australia MenW Working Group. Rise in invasive serogroup W meningococcal disease in Australia 2013-2015. Commun Dis Intell Q Rep. 2016;40:E454–9.PubMedGoogle Scholar
- Tsang RSW, Ahmad T, Tyler S, Lefebvre B, Deeks SL, Gilca R, et al. Whole genome typing of the recently emerged Canadian serogroup W Neisseria meningitidis sequence type 11 clonal complex isolates associated with invasive meningococcal disease. Int J Infect Dis. 2018;69:55–62. DOIPubMedGoogle Scholar
- Potts CC, Joseph SJ, Chang HY, Chen A, Vuong J, Hu F, et al. Population structure of invasive Neisseria meningitidis in the United States, 2011-15. J Infect. 2018;77:427–34. DOIPubMedGoogle Scholar
- Lucidarme J, Hill DMC, Bratcher HB, Gray SJ, du Plessis M, Tsang RSW, et al. Genomic resolution of an aggressive, widespread, diverse and expanding meningococcal serogroup B, C and W lineage. J Infect. 2015;71:544–52. DOIPubMedGoogle Scholar
- Taha MK, Achtman M, Alonso JM, Greenwood B, Ramsay M, Fox A, et al. Serogroup W135 meningococcal disease in Hajj pilgrims. Lancet. 2000;356:2159. DOIPubMedGoogle Scholar
- Weidlich L, Baethgen LF, Mayer LW, Moraes C, Klein CC, Nunes LS, et al. High prevalence of Neisseria meningitidis hypervirulent lineages and emergence of W135:P1.5,2:ST-11 clone in Southern Brazil. J Infect. 2008;57:324–31. DOIPubMedGoogle Scholar
- Lucidarme J, Scott KJ, Ure R, Smith A, Lindsay D, Stenmark B, et al. An international invasive meningococcal disease outbreak due to a novel and rapidly expanding serogroup W strain, Scotland and Sweden, July to August 2015. Euro Surveill. 2016;21:15–23. DOIPubMedGoogle Scholar
- Krone M, Gray S, Abad R, Skoczyńska A, Stefanelli P, van der Ende A, et al. Increase of invasive meningococcal serogroup W disease in Europe, 2013 to 2017. Euro Surveill. 2019;24:24. DOIPubMedGoogle Scholar
- Devoy AF, Dyet KH, Martin DR. Stability of PorA during a meningococcal disease epidemic. J Clin Microbiol. 2005;43:832–7. DOIPubMedGoogle Scholar
- Dyet KH, Martin DR. Clonal analysis of the serogroup B meningococci causing New Zealand’s epidemic. Epidemiol Infect. 2006;134:377–83. DOIPubMedGoogle Scholar
- Sexton K, Lennon D, Oster P, Crengle S, Martin D, Mulholland K, et al. The New Zealand Meningococcal Vaccine Strategy: a tailor-made vaccine to combat a devastating epidemic. N Z Med J. 2004;117:U1015.PubMedGoogle Scholar
- Loring BJ, Turner N, Petousis-Harris H. MeNZB vaccine and epidemic control: when do you stop vaccinating? Vaccine. 2008;26:5899–904. DOIPubMedGoogle Scholar
- Institute of Environmental Science and Research Limited. Notifiable Diseases in New Zealand: Annual Report 2016. Porirua, New Zealand [cited 2020 Nov 3]. https://surv.esr.cri.nz/surveillance/annual_surveillance.php?we_objectID=4656
- Bennett DE, Cafferkey MT. Consecutive use of two multiplex PCR-based assays for simultaneous identification and determination of capsular status of nine common Neisseria meningitidis serogroups associated with invasive disease. J Clin Microbiol. 2006;44:1127–31. DOIPubMedGoogle Scholar
- Oksanen G, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, et al. Vegan: community ecology package [cited 2020 Nov 3]. https://CRAN.R-project.org/package=vegan
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. DOIPubMedGoogle Scholar
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77. DOIPubMedGoogle Scholar
- Kwong JC, Gonçalves da Silva A, Stinear TP, Howden BP, Seemann T. Meningotype: [cited 2020 Nov 3]. https://github.com/MDU-PHL/meningotype
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. DOIPubMedGoogle Scholar
- Okonechnikov K, Conesa A, García-Alcalde F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics. 2016;32:292–4.PubMedGoogle Scholar
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al.; 1000 Genomes Project Analysis Group. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8. DOIPubMedGoogle Scholar
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2. DOIPubMedGoogle Scholar
- Treangen TJ, Ondov BD, Koren S, Phillippy AM. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014;15:524. DOIPubMedGoogle Scholar
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3. DOIPubMedGoogle Scholar
- Felsenstein J. Confidence-limits on phylogenies—an approach using the bootstrap. Evolution. 1985;39:783–91. DOIPubMedGoogle Scholar
- Didelot X, Wilson DJ. ClonalFrameML: efficient inference of recombination in whole bacterial genomes. PLOS Comput Biol. 2015;11:
e1004041 . DOIPubMedGoogle Scholar - Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44(W1):
W242-5 . DOIPubMedGoogle Scholar - Jolley KA, Bray JE, Maiden MCJ. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 2018;3:124. DOIPubMedGoogle Scholar
- Bratcher HB, Corton C, Jolley KA, Parkhill J, Maiden MC. A gene-by-gene population genomics platform: de novo assembly, annotation and genealogical analysis of 108 representative Neisseria meningitidis genomes. BMC Genomics. 2014;15:1138. DOIPubMedGoogle Scholar
- Clinical and Laboratory Standards Institute. Performance standards for antimicrobial susceptibility testing: 28th edition (informational supplement M100). Wayne (PA): The Institute; 2018.
- Zeileis A, Leisch F, Hornik K, Kleiber C, Hansen B, Zeileis MA. Package ‘strucchange’ [cited 2020 Nov 3]. https://CRAN.R-project.org/package=strucchange
- Mowlaboccus S, Jolley KA, Bray JE, Pang S, Lee YT, Bew JD, et al. Clonal expansion of new penicillin-resistant clade of Neisseria meningitidis serogroup W clonal complex 11, Australia. Emerg Infect Dis. 2017;23:1364–7. DOIPubMedGoogle Scholar
- Whaley MJ, Joseph SJ, Retchless AC, Kretz CB, Blain A, Hu F, et al. Whole genome sequencing for investigations of meningococcal outbreaks in the United States: a retrospective analysis. Sci Rep. 2018;8:15803. DOIPubMedGoogle Scholar
- Public Health England. Invasive meningococcal disease in England: annual laboratory confirmed reports for epidemiological year 2017 to 2018. London: Public Health England; 2018 [cited 2020 Nov 3]. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/751821/hpr3818_IMD.pdf
- Knol MJ, de Melker HE, Berbers GAM, van Ravenhorst MB, Ruijs WLM, van Vliet JA. Meningococcal disease in the Netherlands: background information for the Health Council. RIVM Report 2017–0031. 2017 [cited 2020 Nov 03]. https://www.rivm.nl/bibliotheek/rapporten/2017-0031.pdf
- Knol MJ, Hahné SJM, Lucidarme J, Campbell H, de Melker HE, Gray SJ, et al. Temporal associations between national outbreaks of meningococcal serogroup W and C disease in the Netherlands and England: an observational cohort study. Lancet Public Health. 2017;2:e473–82. DOIPubMedGoogle Scholar
- Leimkugel J, Racloz V, Jacintho da Silva L, Pluschke G. Global review of meningococcal disease. A shifting etiology. J Bacterial Res. 2009;1:6–18.
- Bröker M, Jacobsson S, Kuusi M, Pace D, Simões MJ, Skoczynska A, et al. Meningococcal serogroup Y emergence in Europe: update 2011. Hum Vaccin Immunother. 2012;8:1907–11. DOIPubMedGoogle Scholar
- Ministry of Health Manatū Hauora. Change to treatment recommendations for meningococcal disease. 2018 Nov 30 [cited 2020 Nov 3]. https://www.health.govt.nz/news-media/news-items/change-treatment-recommendations-meningococcal-disease
Figures
Cite This ArticleOriginal Publication Date: March 04, 2021
1These authors contributed equally to this article.
Table of Contents – Volume 27, Number 4—April 2021
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Philip E. Carter, Invasive Pathogens Laboratory, Institute of Environmental Science and Research, 34 Kenepuru Dr, Porirua, New Zealand
Top