Volume 28, Number 2—February 2022
Research Letter
Genetic Diversity of Bartonella spp. in Cave-Dwelling Bats and Bat Flies, Costa Rica, 2018
Figure
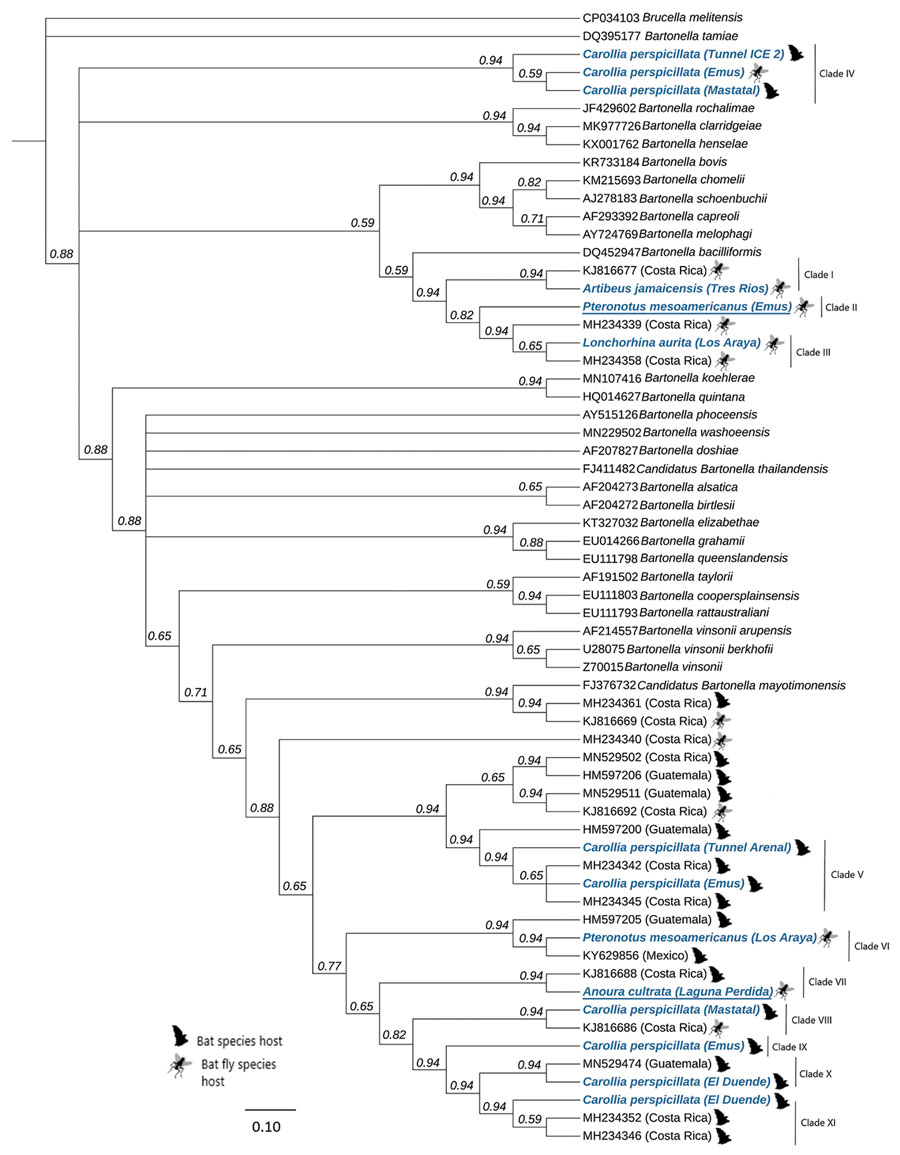
Figure. Phylogenetic tree of 768 bp partial gltA gene of Bartonella variants found in study of Bartonella spp. in bats and bat flies sampled from roost sites, Costa Rica, 2018 (blue), compared with globally named species and other variants found in bats and bat flies in Central America and Mexico. Each sequence is labeled with its GenBank accession number, the organism on which it was detected, and the country of origin. For species in this study, we included the specific site (accession numbers in Appendix Table). Underlining indicates the potential newly described genotypes. We constructed the global phylogenetic tree by using Bayesian Markov chain Monte Carlo (MrBayes 2.2.4, https://www.geneious.com), with 1 million generations and a burn-in fraction of 25%. We determined the parameters for the nucleotide changes by using MEGA X (https://mafft.cbrc.jp/alignment/software). Inner node labels identify consensus support. Scale bar indicates nucleotide substitutions/site (%).
1These authors contributed equally to this article.