Volume 28, Number 5—May 2022
Dispatch
Novel Hendra Virus Variant Circulating in Black Flying Foxes and Grey-Headed Flying Foxes, Australia
Cite This Article
Citation for Media
Abstract
A novel Hendra virus variant, genotype 2, was recently discovered in a horse that died after acute illness and in Pteropus flying fox tissues in Australia. We detected the variant in flying fox urine, the pathway relevant for spillover, supporting an expanded geographic range of Hendra virus risk to horses and humans.
Hendra virus (HeV; genus Henipavirus, family Paramyxoviridae) is a well-characterised zoonotic pathogen endemic to Pteropus spp. bats (flying foxes) in Australia. Spillover from bats to horses has been detected 63 times; 4 of 7 persons infected from horses have died (1). Quantitative reverse-transcription PCR (qRT-PCR) (2) is a tool used for surveillance and priority disease investigation in bats and horses (3,4). The high specificity of assays limits detection to a narrow range of genotypic diversity, meaning that divergent variants might remain undetected (3).
In October 2021, spillover of a novel variant, HeV genotype 2 (HeV-g2), resulted in the death of a horse in New South Wales (NSW), Australia, farther south than HeV had previously been detected in horses (5). This spillover was detected only because diagnostic assays had been recently updated after retrospective discovery of HeV-g2 in a horse that exhibited signs of HeV disease in 2015 but tested negative through routine screening at that time (3). Discovery of HeV-g2 in this horse arose using broad panparamyxovirus PCRs (6), followed by next-generation sequencing and virus isolation. The variant showed 84% pairwise nucleotide identity genomewide to prototype HeV (HeV-g1), and 99% similarity with partial sequences recovered from tissue samples from a grey-headed flying fox, P. poliocephalus (7). Bats submitted for lyssavirus diagnostics were opportunistically screened using an updated quantitative PCR specific for HeV-g2, which resulted in additional positive detections in tissue collected from P. poliocephalus in 2019–2021 and a little red flying fox (P. scapulatus) in 2015 (7).
Although HeV-g1 has been detected in tissues from all 4 flying fox species in continental Australia, excretion of the virus has been confirmed only in the black flying fox (P. alecto) and the spectacled flying fox (P. conspicillatus), suggesting these species are sources of transmission to horses (8,9). Sequence mismatches between HeV-g1 and HeV-g2 mean that PCR assays used in previous surveillance of reservoir hosts would not have detected the novel HeV-g2. To address this gap, we used a new qRT-PCR (3) to screen banked flying fox urine samples collected over a large extent of space and time.
Figure
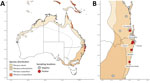
We collected pooled urine samples from plastic sheets placed underneath flying fox roosts in southeastern Queensland and mid- to north-coast NSW during December 2016–September 2020 (Figure). We placed sheets in areas of the roost where P. alecto flying foxes were roosting, although other species were often also present. We recorded the number and species of bats immediately above the sheets. We also captured individual bats in mist nests; recorded species, sex, and age class; then collected urine samples directly from each anaesthetised bat or from a urine collection bag attached to its holding bag. Shortly after collection, we placed samples into viral lysis buffer, virus transport media, or an empty cryovial and stored them at −80°C (Appendix).
We used the QIAamp Viral RNA Kit using a QIAcube HT automated system (QIAGEN, https://www.qiagen.com) to extract RNA, then eluted it in 150 μL of TE buffer and first screened it for HeV-g1 using a qRT-PCR assay targeting the P gene (Table 1). We stored extracted RNA at −80°C and then screened it for HeV-g2 using the new multiplexed qRT-PCR assay, targeting the M gene with primers specific for HeV-g1 and HeV-g2 (2,3) (Table 1; Appendix). We used 10-fold dilutions with a known number of genome copies to construct a standard curve, calculate copy numbers/mL, and estimate limit of detection. We amplified the partial cytochrome b gene from all positive samples (10,11) (Table 1) and confirmed host species identity based on sequence identity across 402-bp sequences (Appendix).
We screened 4,539 pooled urine samples collected from 129 underroost sampling sessions and 1,674 urine samples collected from individual bats over 39 catching sessions during July 2017–September 2020 (Appendix Tables 1, 2). Eight pooled urine samples and 2 samples from individual flying foxes tested positive for HeV-g2 (Table 2). Positive samples were from Sunnybank in Queensland and Clunes, Lismore, Dorroughby, Maclean, and Nambucca Heads in NSW.
We detected HeV-g2 in samples collected across all seasons. Prevalence in sessions with positive detections ranged from 2.5% to 6.5% (95% CI 0.1%–22.8%). In pooled samples, HeV-g2 was only detected in sessions when HeV-g1 was also detected (HeV-g1 prevalence range 2.5%–50.1%); however, we found no statistically significant correlation between HeV-g1 and HeV-g2 prevalence (Pearson correlation analysis ρ = 0.09; p = 0.87). Most (8/10) of the HeV-g2–positive samples had low genome copies, but 2, ARSUN015_15_1 and ARLIS002_55_1, had considerably higher copy numbers (Table 2).
Individual flying foxes that tested positive included a P. poliocephalus juvenile female captured in Maclean, NSW, and a P. alecto adult male captured in Clunes, NSW (Appendix Table 3). We detected HeV-g2 in pooled samples from mixed-species roosts containing P. alecto and P. poliocephalus flying foxes. Cytochrome b sequencing identified DNA from P. alecto flying foxes in 6/8 positive underroost samples and from P. poliocephalus flying foxes in 2/8 (Table 2).
Urine is the route of HeV excretion from flying foxes and the source of virus transmission to horses. Detecting the novel Hendra variant HeV-g2 in the urine of flying foxes helped identify its distribution range, associated host species, transmission dynamics, and spillover risk. We show evidence that P. alecto and P. poliocephalus flying foxes excrete HeV-g2 in urine and both are likely competent reservoir hosts. We did not screen urine samples from P. conspicillatus or P. scapulatus flying foxes, so the potential of these species to excrete HeV-g2 in urine remains unconfirmed.
Although HeV-g1 has been detected in flying fox urine samples collected across all seasons, prevalence peaks in winter in subtropical regions (4,12), which is consistent with our preliminary HeV-g2 seasonality findings (5/8 detections in late May–late August) in the study area. The significantly lower prevalence of HeV-g2 than HeV-g1 could indicate actual lower prevalence in the sampled population. Alternatively, repeated freeze-thaw cycles in our samples or the bias toward collecting P. alecto urine in our sampling design might have led to lower detection. Tissue samples from flying foxes submitted for lyssavirus testing after contact with humans or pets showed higher HeV-g2 prevalence than our samples from wild populations (7), which might reflect higher prevalence in sick or stressed bats or geographical differences. HeV-g2 was previously detected in tissue samples from South Australia (3 positives from 4 samples), Victoria (7/64), and Western Australia (1/2) (7). Our findings extend the known distributional range of HeV-g2 to southeastern Queensland and mid- to north-coast NSW, areas proximate to the 2 known cases of HeV-g2 spillover to horses (3,5).
Our findings support expanding the expected geographic risk area for HeV spillover to include the distribution of P. poliocephalus flying foxes. Screening flying fox urine samples from a broader geographic range, including regions where P. alecto flying foxes are absent, should better inform epidemiologic relationships and relative prevalence of HeV variants. Given that data on the true diversity of HeV and related viruses in flying fox populations are incomplete, unbiased or Paramyxoviridae family–level viral surveillance in reservoir and spillover hosts might identify further variants. Developing a panel of diagnostic tools to detect a more comprehensive range of the viruses capable of spillover would substantially advance our ability to forecast spillover risk, manage biosecurity, and provide guidance to horse owners, veterinarians, and other stakeholders.
Dr. Peel is a DECRA senior research fellow at Griffith University in Brisbane, Queensland, Australia. Her primary interests lie in the role of landscape change and anthropogenic influence on the dynamics and drivers of infectious disease in bats. Dr. Yinda is a postdoctoral research fellow at the Virus Ecology Section of the Rocky Mountains Laboratories, National Institute of Allergy and Infectious Diseases, National Institutes of Health. He is interested in pathogen discovery of emerging viruses, and their cross-species transmissions and disease potential.
Bat One Health group members: Mandy Allonby, Remy Brooks, Liam Chirio, Caylee A. Falvo, Hamish McCallum, Ticha Padgett-Stewart, Manuel Ruiz-Aravena, Kirk A. Silas, and Rachael Smethurst.
Acknowledgments
We acknowledge the Bundjalung, Butchulla, Danggan Balun, Gomeroi, Gumbainggir, Kabi Kabi, Taribelang Bunda, Turrbal, Widjabul Wia-bal, Yugambeh, and Yuggera Ugarapul people, who are the traditional custodians of the land upon which this work was conducted. We also thank government and private landholders for granting permission for fieldwork and broader team members and volunteers for their contributions: Liam McGuire, Wyatt Madden, Justine Scaccia, Denise Karkkainen, Cara Parsons, Ariane Ananda, Emma Glennon, Emily Stanford, Jessica Mitchell, Eloise Stephenson, Kerryn Parry-Jones, Anja Divljan, Cinthia Pietromonaco, and other volunteers. We thank Allan Grolla for the design of the HeV assay and Paul Oliver for input on interpretation of cytochrome b sequencing data.
The project was supported by an NSF Coupled Dynamics of Natural and Human Systems grant DEB1716698, funding from the Defense Advanced Research Projects Agency administered through Cooperative Agreement D18AC00031-PREEMPT and support from the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health. A.J.P. was supported by an ARC DECRA fellowship (DE190100710). R.K.P. was supported by the USDA National Institute of Food and Agriculture (Hatch project 1015891). E.J.A., J.S.E., and I.L.S. were supported by the Australian Government Department of Agriculture, Water and the Environment, Biosecurity Innovation Project 2020–21 Project ID 202043, Metagenomic Investigation of Horses as Sentinels.
References
- Playford EG, McCall B, Smith G, Slinko V, Allen G, Smith I, et al. Human Hendra virus encephalitis associated with equine outbreak, Australia, 2008. Emerg Infect Dis. 2010;16:219–23. DOIPubMedGoogle Scholar
- Smith IL, Halpin K, Warrilow D, Smith GA. Development of a fluorogenic RT-PCR assay (TaqMan) for the detection of Hendra virus. J Virol Methods. 2001;98:33–40. DOIPubMedGoogle Scholar
- Annand EJ, Horsburgh BA, Xu K, Reid PA, Poole B, de Kantzow MC, et al. Novel Hendra virus variant detected by sentinel surveillance of Australian horses. Emerg Infect Dis. 2022;28:693–704. DOIPubMedGoogle Scholar
- Field H, Jordan D, Edson D, Morris S, Melville D, Parry-Jones K, et al. Spatiotemporal aspects of Hendra virus infection in pteropid bats (flying-foxes) in eastern Australia. PLoS One. 2015;10:
e0144055 . DOIPubMedGoogle Scholar - Tong S, Chern S-WW, Li Y, Pallansch MA, Anderson LJ. Sensitive and broadly reactive reverse transcription-PCR assays to detect novel paramyxoviruses. J Clin Microbiol. 2008;46:2652–8. DOIPubMedGoogle Scholar
- Wang J, Anderson DE, Halpin K, Hong X, Chen H, Walker S, et al. A new Hendra virus genotype found in Australian flying foxes. Virol J. 2021;18:197. DOIPubMedGoogle Scholar
- Edson D, Field H, McMichael L, Vidgen M, Goldspink L, Broos A, et al. Routes of Hendra virus excretion in naturally-infected flying-foxes: implications for viral transmission and spillover risk. PLoS One. 2015;10:
e0140670 . DOIPubMedGoogle Scholar - Smith I, Broos A, de Jong C, Zeddeman A, Smith C, Smith G, et al. Identifying Hendra virus diversity in pteropid bats. PLoS One. 2011;6:
e25275 . DOIPubMedGoogle Scholar - Kocher TD, Thomas WK, Meyer A, Edwards SV, Pääbo S, Villablanca FX, et al. Dynamics of mitochondrial DNA evolution in animals: amplification and sequencing with conserved primers. Proc Natl Acad Sci U S A. 1989;86:6196–200. DOIPubMedGoogle Scholar
- Hsieh H-M, Chiang H-L, Tsai L-C, Lai S-Y, Huang N-E, Linacre A, et al. Cytochrome b gene for species identification of the conservation animals. Forensic Sci Int. 2001;122:7–18. DOIPubMedGoogle Scholar
- Plowright RK, Eby P, Hudson PJ, Smith IL, Westcott D, Bryden WL, et al. Ecological dynamics of emerging bat virus spillover. Proc Biol Sci. 2015;282:
20142124 . DOIPubMedGoogle Scholar - Lunney D, Richards G. Dickman C Pteropus poliocephalus. The IUCN Red List of Threatened Species [cited 2021 Nov 15]. https://www.iucnredlist.org/species/18751/22085511
Figure
Tables
Cite This ArticleOriginal Publication Date: April 07, 2022
1These authors contributed equally to this article.
2These senior authors contributed equally to this article.
3Members of Bat One Health are listed at the end of this article.
Table of Contents – Volume 28, Number 5—May 2022
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Alison Peel, Centre for Planetary Health and Food Security, Griffith University, Nathan Campus, Sir Samuel Griffith Centre (N78) 2.23, 170 Kessels Rd, Nathan, QLD 4111, Australia
Top