Fatal Human Neurologic Infection Caused by Pigeon Avian Paramyxovirus-1, Australia
Siobhan Hurley
1
, John Sebastian Eden
1, John Bingham, Michael Rodriguez, Matthew J. Neave, Alexandra Johnson, Annaleise R. Howard-Jones, Jen Kok, Antoinette Anazodo, Brendan McMullan, David T. Williams, James Watson, Annalisa Solinas, Ki Wook Kim
2, and William Rawlinson
2
Author affiliations: Prince of Wales Hospital, Randwick, New South Wales, Australia (S. Hurley, K.W. Kim); Westmead Institute for Medical Research Centre for Virus Research, Westmead, New South Wales, Australia (J.S. Eden); Sydney Institute for Infectious Diseases, University of Sydney Faculty of Medicine and Health, Sydney, New South Wales, Australia (J.S. Eden, A.R. Howard-Jones); CSIRO Australian Centre for Disease Preparedness, Geelong, Victoria, Australia (J. Bingham, M.J. Neave, D.T. Williams, J. Watson); Prince of Wales and Sydney Children’s Hospital, Randwick (M. Rodriguez, A. Solinas); Sydney Children’s Hospital, Randwick (A. Johnson, B. McMullan); Centre for Infectious Diseases and Microbiology Laboratory Services, New South Wales Health Pathology–Institute of Clinical Pathology and Medical Research, Westmead (A.R. Howard-Jones, J. Kok); Kids Cancer Centre, Sydney Children’s Hospital, Randwick (A. Anazodo); University of New South Wales Faculty of Medicine and Health, School of Clinical Medicine, Sydney (B. McMullan, K. Kim); Prince of Wales Hospital and Community Health Services, Sydney (W. Rawlinson); University of New South Wales Schools of Clinical Medicine, Biotechnology and Biomolecular Sciences, Sydney (W. Rawlinson)
Main Article
Figure 5
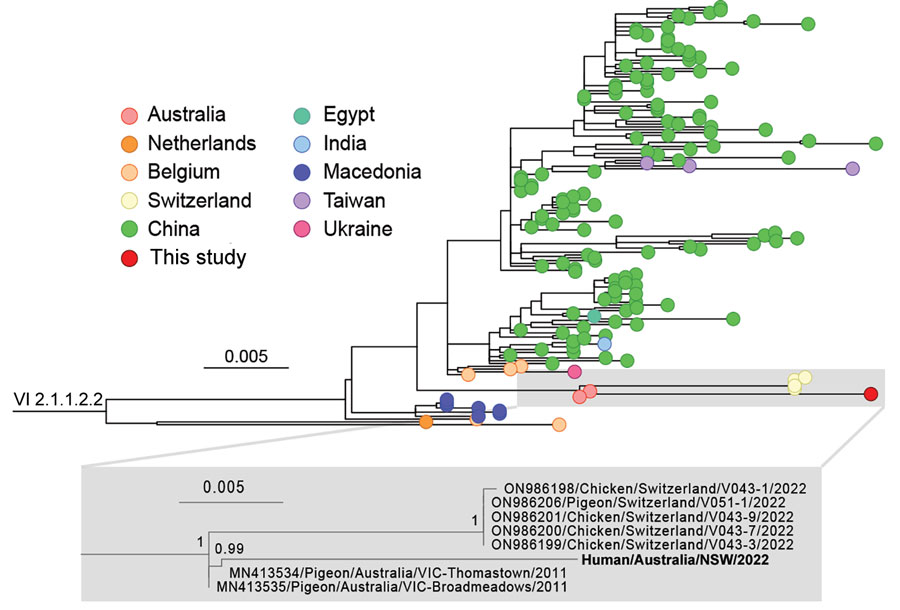
Figure 5. Phylogeny of avian paramyxovirus type 1 strain from an immunocompromised child in Australia (bold). Tree was prepared from MAFFT-aligned fusion gene sequences of genotype VI sublineage 2.1.1.2.2 strains (classified as pigeon avian paramyxovirus 1) using PhyML with the Hasegawa-Kishino-Yano + gamma DNA substitution model and rooted with a genotype VI sublineage 2.1.1.2.1 outgroup (HM063425/Pigeon/CHN/P4). Red dot indicates virus from this case; GenBank accession numbers are provided for reference sequences. Gray box indicates the branch containing the virus we identified, expanded to show detail. Colored circles at tips indicate country of sampling. The virus from our study appears to be related to viruses circulating in Australia since at least 2011. Node support values show Shimodaira-Hasegawa—like approximate likelihood ratio test statistics; branch lengths are proportional to the scale of the number of substitutions per site. Scale bars indicate number of substitutions per site. All strains used in this analysis are listed in the Appendix.
Main Article
Page created: November 13, 2023
Page updated: November 18, 2023
Page reviewed: November 18, 2023
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.