Volume 29, Number 7—July 2023
Research Letter
Novel Highly Pathogenic Avian Influenza A(H5N1) Clade 2.3.4.4b Virus in Wild Birds, South Korea
Cite This Article
Citation for Media
Abstract
We isolated 5 highly pathogenic avian influenza A(H5N1) clade 2.3.4.4.b viruses from wild waterfowl feces in South Korea during November 2022. Whole-genome sequencing and phylogenetic analysis revealed novel genotypes produced by reassortment with Eurasian low pathogenicity avian influenza viruses. Enhanced surveillance will be required to improve prevention and control strategies.
Highly pathogenic avian influenza viruses (HPAIVs) have caused major economic losses in the poultry industry and are a major threat to public health. Since the first detection of HPAIV A(H5N1) from a goose in 1996 in Guangdong, China, its descendants have evolved into multiple hemagglutinin (HA) gene-specific clades (H0–H9) and subclades (1) causing intercontinental epizootics (2). Over several decades, H5 HPAIVs have evolved into multiple subtypes and genotypes generated by reassortment with low pathogenicity avian influenza viruses (LPAIVs), which led to emergence of clade 2.3.4.4 H5Nx HPAIVs in eastern China during 2013–2014 (3).
In mid-2016, reassortant H5N8 clade 2.3.4.4b HPAIVs that contained internal genes of LPAIVs from Eurasia were detected in wild birds at Uvs-Nuur Lake in Russia and Qinghai Lake in China (4); the viruses caused large outbreaks in Europe during 2016–2017 (5). Subsequently, various novel reassortant H5N8 HPAIVs were detected in Eurasia (5,6). In late 2020, novel reassortant clade 2.3.4.4b H5N1 HPAIVs were detected and became predominant in Europe in poultry and wild birds (5).
We isolated 5 H5N1 HPAIVs from wild bird feces collected in South Korea in November 2022 (Appendix 1): A/Spot-billed_duck/Korea/K22-730-1/2022(H5N1) [K22-730-1], A/Wild_bird/Korea/K22-742/2022(H5N1) [K22-742], A/Spot-billed_duck/Korea/K22-856-2/2022(H5N1) [K22-856-2], A/Spot-billed_duck/Korea/K22-862-1/2022(H5N1) [K22-862-1], and A/Spot-billed_duck/Korea/K22-920/2022(H5N1) [K22-920] (Appendix 1 Table 1). To rapidly share the information, we conducted whole-genome sequencing of the isolates and deposited the genome sequences in the GISAID database (https://www.gisaid.org).
All H5N1 isolates were classified as HPAIV on the basis of HA cleavage site amino acid sequences (PLRPKRRKR/G). The 5 isolates shared high nucleotide sequence identities (99.4% to ≈100%) across all 8 influenza genes, except for the K22-920 isolate polymerase basic protein 1 (PB1), polymerase acidic protein (PA), nucleoprotein (NP), and nonstructural (NS) genes (93.0% to ≈99.0%). BLAST (https://blast.ncbi.nlm.nih.gov) search results showed HA, neuraminidase, and matrix (M) protein genes of all isolates had >99.1% identities with 2021–2022 clade 2.3.4.4b HPAIVs (Table 1). PB1, PA, NP, and NS genes of all isolates were highly similar (98.72%–99.52%) to 2019–2022 LPAIVs from East Asia. PB1, PA, NP, and NS genes of K22-920 were similar to 2019–2020 LPAIVs from South Korea, Russia, and Bangladesh (>98.4%–99.3%).
Figure 1
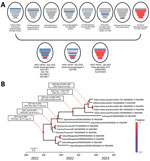
Figure 1. Phylogenetic analysis of novel highly pathogenic avian influenza A(H5N1) clade 2.3.4.4b viruses found in wild bird feces in South Korea, November 2022. A) Schematic representation of origin of virus isolates...
In maximum-likelihood phylogenetic analyses, PB2, HA, neuraminidase, and M genes of the 5 H5N1 viruses from South Korea clustered with those of viruses previously described as genotype G10, identified in China during 2022–2023 (Appendix 1 Figures 1–8); G10 is a natural reassortant H5N1 HPAIV containing the PB2 gene from LPAIVs (7). PB1, PA, NP, and NS genes of all H5N1 viruses from South Korea except K22-920 clustered with those of LPAIVs from Asia; those gene segments in K22-920 clustered separately with other LPAIVs from Asia, including South Korea, Russia, and Bangladesh (Appendix 1 Figures 2, 3, 5, 8). Bayesian phylogeny of the HA gene indicated the H5N1 viruses from South Korea formed a well-supported cluster; time to most recent common ancestor was estimated to be August 11, 2022 (95% highest posterior density June 11–October 11, 2022), suggesting those H5N1 HPAIVs most likely emerged 1–2 months before the autumn wild bird migration to South Korea (Figure 1; Appendix 1 Figure 9). The isolates from South Korea shared recent common ancestry with the A/Jiangsu/NJ210/2023(H5N1) virus; time to most recent common ancestor between them was April 12, 2022 (95% highest posterior density December 26, 2021–July 28, 2022), suggesting the ancestral H5N1 HPAIVs had been circulating undetected for ≈7 months.
The H5N1 HPAIVs from South Korea contained amino acids in HA with binding affinity for avian α-2,3-linked sialic acid receptors (T118, V210, Q222, and G224) (H5 numbering) (8,9). They also had 2 HA amino acid substitutions, S113A and T156A, associated with increased binding affinity to human α-2,6-linked sialic acid receptors (Appendix 1 Table 2). All 5 isolates had amino acid substitutions that included A515T in PA, known to increase polymerase activity in mammal cells, and N30D, I43M, T215A in MP1 and L89V in PB2, known to increase virulence in mice (Appendix 1 Tables 2, 3).
The HPAI/LPAI reassortment of H5Nx clade 2.3.4.4b HPAIVs created a diverse genetic pool of H5 clade 2.3.4.4 viruses that continuously emerged in various countries (1). Clade 2.3.4.4 H5N8 HPAIV isolated from Uvs-Nuur Lake in Russia had reassorted H3N8 LPAIV genes from Mongolia (4). In Europe, HPAIVs identified in 2020 (5,6) were produced by reassortment between clade 2.3.4.4b HPAIV and LPAIVs from Eurasia. Novel reassortments of clade 2.3.4.4 HPAIV and LPAIVs from Eurasia were also detected in 2016 (10), during 2020–2021 (Appendix 1 reference 1), and in late 2021 (Appendix 1 reference 2) in South Korea. Considering the continuous emergence and global dissemination of novel reassortant clade 2.3.4.4b HPAI H5Nx viruses, enhanced active surveillance in wild animals and domestic poultry will be required to monitor the introduction, dissemination, and evolution of HPAIVs and provide insight for improved prevention and control strategies.
Mr. Lee is a PhD candidate at Konkuk University, Seoul, South Korea. His primary research interests focus on molecular epidemiology and host–pathogen interactions of avian influenza viruses.
Acknowledgments
We thank our colleagues worldwide for their laboratory contributions, which are made available through GISAID (Appendix 2).
This research was supported by the Bio and Medical Technology Development Program of the National Research Foundation, funded by the government of South Korea (grant no. NRF-2018M3A9H405635), and by the National Institute of Wildlife Disease Control and Prevention, South Korea (grant no. 2022-007).
References
- Gu M, Liu W, Cao Y, Peng D, Wang X, Wan H, et al. Novel reassortant highly pathogenic avian influenza (H5N5) viruses in domestic ducks, China. Emerg Infect Dis. 2011;17:1060–3. DOIPubMedGoogle Scholar
- Lee DH, Criado MF, Swayne DE. Pathobiological origins and evolutionary history of highly pathogenic avian influenza viruses. Cold Spring Harb Perspect Med. 2021;11:
a038679 . DOIPubMedGoogle Scholar - Lee Y-J, Kang H-M, Lee E-K, Song B-M, Jeong J, Kwon Y-K, et al. Novel reassortant influenza A(H5N8) viruses, South Korea, 2014. Emerg Infect Dis. 2014;20:1087–9. DOIPubMedGoogle Scholar
- Lee DH, Sharshov K, Swayne DE, Kurskaya O, Sobolev I, Kabilov M, et al. Novel reassortant clade 2.3.4.4 avian influenza A(H5N8) virus in wild aquatic birds, Russia, 2016. Emerg Infect Dis. 2017;23:359–60. DOIPubMedGoogle Scholar
- Pohlmann A, King J, Fusaro A, Zecchin B, Banyard AC, Brown IH, et al. Has epizootic become enzootic? Evidence for a fundamental change in the infection dynamics of highly pathogenic avian influenza in Europe, 2021. MBio. 2022;13:
e0060922 . DOIPubMedGoogle Scholar - Lewis NS, Banyard AC, Whittard E, Karibayev T, Al Kafagi T, Chvala I, et al. Emergence and spread of novel H5N8, H5N5 and H5N1 clade 2.3.4.4 highly pathogenic avian influenza in 2020. Emerg Microbes Infect. 2021;10:148–51. DOIPubMedGoogle Scholar
- Cui P, Shi J, Wang C, Zhang Y, Xing X, Kong H, et al. Global dissemination of H5N1 influenza viruses bearing the clade 2.3.4.4b HA gene and biologic analysis of the ones detected in China. Emerg Microbes Infect. 2022;11:1693–704. DOIPubMedGoogle Scholar
- Burke DF, Smith DJ. A recommended numbering scheme for influenza A HA subtypes. PLoS One. 2014;9:
e112302 . DOIPubMedGoogle Scholar - Suttie A, Deng YM, Greenhill AR, Dussart P, Horwood PF, Karlsson EA. Inventory of molecular markers affecting biological characteristics of avian influenza A viruses. Virus Genes. 2019;55:739–68. DOIPubMedGoogle Scholar
- Kwon JH, Lee DH, Swayne DE, Noh JY, Yuk SS, Erdene-Ochir TO, et al. Reassortant clade 2.3.4.4 avian influenza A(H5N6) virus in a wild mandarin duck, South Korea, 2016. Emerg Infect Dis. 2017;23:822–6. DOIPubMedGoogle Scholar
Figure
Table
Cite This ArticleOriginal Publication Date: May 19, 2023
Table of Contents – Volume 29, Number 7—July 2023
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Dong-Hun Lee, Wildlife Health Laboratory, College of Veterinary Medicine, Konkuk University, Seoul, South Korea
Top