Volume 3, Number 4—December 1997
THEME ISSUE
Foodborne
Special Issue
Infectious Disease as an Evolutionary Paradigm
Cite This Article
Citation for Media
The basic principles of genetics and evolution apply equally to human hosts and to emerging infections, in which foodborne outbreaks play an important and growing role. However, we are dealing with a very complicated coevolutionary process in which infectious agent outcomes range from mutual annihilation to mutual integration and resynthesis of a new species. In our race against microbial evolution, new molecular biology tools will help us study the past; education and a global public health perspective will help us deal better with the future.
Figure 1
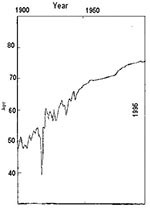
Figure 1. Life expectancy in the United States, at birth, 20th century.
Figure 2

Figure 2. Trends in infectious diseases mortality, 1900-1992. Source: CDC, unpub. data.
Life expectancy in the United States from 1900 to the present (Figure 1) shows an overall steady rise, reflecting improved health conditions in general, the result of advances in medical science, hygiene, personal care, health technologies, and public health administrations. The rise decelerates asymptotically to a near plateau from the 1950s to the 1970s, reflecting an epidemic of coronary disease, which we do not yet fully understand. Improvements in medical care, attention to life style, or indiscriminate use of aspirin may all be responsible for the subsequent decrease in deaths from coronary disease. Up to the 1940s, the rising curve is jagged, reflecting sporadic infectious disease outbreaks, especially the Spanish influenza outbreak of 1918. Whether the life expectancy curve continues to rise smoothly or whether it has some jagged declines depends on what we do about transmission of infectious disease, including foodborne disease. When plotted another way (Figure 2), both the absolute number of deaths from infectious disease and the proportion of total deaths attributable to infectious disease also show steady amelioration from 1900 almost to the present.
Figure 3

Figure 3. Pneumonia and influenza mortality, by age, in certain epidemic years. (Reprinted with permission of W. Paul Glezen and Epidemiologic Reviews. Emerging Infections: Pandemic Influenza. Epi Rev 1996;18:66).
The 1918 Spanish influenza pandemic may be a prototype for future emerging infections. Although minimized as not much more than a bad cold, influenza took a terrible toll in 1918, especially on young people (Figure 3).
Somewhat older persons may have been protected by immunity from prior exposure to related strains of influenza. The disease, with rapid onset of fulminating pneumonic symptoms, killed 20 to 25 million persons worldwide. The infectious agent was not available for study at that time. However, very recently the Armed Forces Institute of Pathology recovered with PCR technology genetic fragments of the 1918 influenza virus (1). Less than 10% of the entire genome has been recovered to date, but recovery of complete sequences is likely. Although the target genes have not yet provided a clue as to why the 1918 influenza was so devastating, they demonstrate the enormous potential of today's molecular biology tools.
These tools will enable us to better study paleovirology and paleomicrobiology. We are accustomed to stereotyping historical disease outbreaks as if we really knew what they were, but we really know very little detail about their genetic features. For example, we talk about the great historic plagues as if they indeed were Yersinia or cholera or malaria. We should look forward to finding out about the 14th century black death, if it was indeed Yersinia pestis. Although clinically unmistakable, that is not to say it was caused by the identical genotype of present Yersinia strains.
We need to look ahead as well as back. In this century, emerging and reemerging infections have stimulated flurries of interest, but in general we have been complacent about infectious diseases ever since the introduction of antibiotics. The effect of antibiotics on acute infections and tuberculosis as well as the effect of polio vaccination led to a national, almost worldwide, redirection of attention to chronic and constitutional diseases. However, the HIV pandemic in the early 1980s caught us off guard, reminding us that there are many more infectious agents in the world. It is fortuitous that retroviruses had already been studied from the perspective of cancer etiology; otherwise, we would have had no scientific platform whatsoever for coping with HIV and AIDS.
The Committee on International Science Engineering and Technology provided an interagency review setting out a policy framework for the United States' global response to infectious disease (Table 1). The policy provides a worldwide mantle for surveillance and monitoring, remedial measures, development of new drugs, vaccines, and treatment modalities. The global outlook is necessary, even if for purely selfish reasons, because to infectious agents the world is indivisible, with no national boundaries. Our thinking has been impoverished in terms of budget allocations for dealing with health on an international basis.
We are engaged in a type of race, enmeshing our ecologic circumstances with evolutionary changes in our predatory competitors. To our advantage, we have wonderful new technology; we have rising life expectancy curves. To our disadvantage, we have crowding; we have social, political, economic, and hygienic stratification. We have crowded together a hotbed of opportunity for infectious agents to spread over a significant part of the population. Affluent and mobile people are ready, willing, and able to carry afflictions all over the world within 24 hours' notice. This condensation, stratification, and mobility is unique, defining us as a very different species from what we were 100 years ago. We are enabled by a different set of technologies. But despite many potential defenses—vaccines, antibiotics, diagnostic tools—we are intrinsically more vulnerable than before, at least in terms of pandemic and communicable diseases.
We could imaginably adapt in a Darwinian fashion, but the odds are stacked against us. We cannot compete with microorganisms whose populations are measured in exponents of 1012, 1014, 1016 over periods of days. Darwinian natural selection has led to the evolution of our species but at a terrible cost. If we were to rely strictly on biologic selection to respond to the selective factors of infectious disease, the population would fluctuate from billions down to perhaps millions before slowly rising again. Therefore, our evolutionary capability may be dismissed as almost totally inconsequential. In the race against microbial genes, our best weapon is our wits, not natural selection on our genes.
New mechanisms of genetic plasticity of one microbe species or another are uncovered almost daily. Spontaneous mutation is just the beginning. We are also dealing with very large populations, living in a sea of mutagenic influences (e.g., sunlight). Haploid microbes can immediately express their genetic variations. They have a wide range of repair mechanisms, themselves subject to genetic control. Some strains are highly mutable by not repairing their DNA; others are relatively more stable. They are extraordinarily flexible in responding to environmental stresses (e.g., pathogens' responses to antibodies, saprophytes' responses to new environments). Mechanisms proliferate whereby bacteria and viruses exchange genetic material quite promiscuously. Plasmids now spread throughout the microbial world (3). They can cross the boundaries of yeast and bacteria. Lateral transfer is very important in the evolution of microorganisms. Their pathogenicity, their toxicity, their antibiotic resistance do not rely exclusively on evolution within a single clonal proliferation.
We have a very powerful theoretical basis whereby the application of selective pressure (e.g., antibiotics in food animals) will result in drug resistance carried by plasmids, or pathogens attacking humans. It is not easy to get direct and immediate epidemiologic evidence, but the foundations for these phenomena exist and must be taken into account in the development of policies. We have barely begun to study the responses of microorganisms under stress, although we have examples where root mechanisms of adaptive mutability are themselves responses to stress. In recent experiments, bacterial restriction systems are more permissive of the introduction of foreign DNA, possibly letting down their guard in response to "mutate or die" circumstances. This does not reflect bacterial intelligence—that they know exactly what mutations they should undergo in response to environmental situations. Their intrinsic mutability and capacity to exchange genetic information without knowing what it is going to be is not a constant; it is certainly under genetic control and in some circumstances varies with the stress under which the microbes are placed.
Figure 4
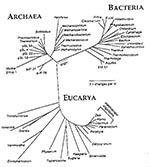
Figure 4. The three-domain tree of life based on small-subunit rRNA sequences. Reprinted with permission of Norman R. Pace and ASM News. ASM News 1996;62 (9):464.
Evolution is more or less proportionate to the degree of genetic divergence among the different branches of the three-tiered tree of life, with the archaeal branch, the eubacterial branch, and the eukaryotes (Figure 4). The tree illustrates the small territory occupied by humans in the overall world of biodiversity. It shows mitochondria right next to Escherichia coli. Bacterial invasion of a primitive eukaryote 2-1/2 to 3 billion years ago, synchronized with the development of primitive green oxygen-generating plants, conferred a selective advantage to complexes that could use oxygen in respiration. Our ancestors were once invaded by an oxidative-capable bacterium that we now call a mitochondrium and that is present in every cell of everybody and almost every species of eukaryote. We did not evolve in a monotonous treelike development; we are also the resynthesis of components of genetic development that diverged as far as the bacteria and were reincorporated into the mitochondrial part of our overall genome. Another example of lateral transfer is the symbiosis that resulted from chloroplast invasion of green plants.
The outcome of encounters between mutually antagonistic organisms is intrinsically unpredictable. The 1918 influenza outbreak killed half percent of the human population; but because the consequences were to either kill the host or leave the host immune, the virus died out totally, leaving no trace in our genomes, as far as we know. Historic serology on survivors has found memory cells and antibodies against H1N1, the serotype of the resurrected 1918 virus. Unlike the influenza virus, which left no known genetic imprint, 400 to 500 retroviruses are integrated into our human genome. The full phylogeny of these encounters is unknown, but many of these viruses may precede the separation of homo sapiens from the rest of the hominid line.
Infectious agent outcomes range from mutual annihilation to mutual integration and resynthesis of a new species. Much has been made of the fact that zoonoses are often more lethal to humans than to their original host, but this phenomenon cannot necessarily be generalized. Most zoonoses do not affect humans adversely. Some are equally capable in a new host. We tend to pay most attention, however, to those, such as yellow fever, for which we have not genetically or serologically adapted and which cause severe disease.
Canine distemper provides an example of a quasihereditary adaptation. In the Serengeti, the disease migrated from village dogs to jackals, which shared prey and had contact with lions. About one-fourth of the preserve's 4,000 lions died of canine distemper (4) but the survivors are immune and will pass immunoglobulin, to their offspring. The cubs' maternal immunity will likely mitigate infection and permit a new equilibrium, not because of genetic adaptation but because of the preimmunized host. This is also the most plausible explanation for how savage the polio virus has been as a paralytic infection of young people. It may also apply to hepatitis, where cleaner is not always better if it means we do not have the "street smarts" to respond to new infectious challenges. These nongenetic adaptations between parasite and host complicate our outcome expectations.
Short-term shifts in equilibrium can give ferocious but temporary advantages to a virus. Long-term outcomes are most stable when they involve some degree of mutual accommodation, with both surviving longer. New short-term deviants, however, can disrupt this equilibrium. The final outcome of the HIV pandemic cannot be predicted. More strains with longer latency may be taking over, mitigating the disease. However, deviant strains could counteract this effect by overcoming immunity and rapidly proliferating, with earlier and more lethal consequences.
We should also consider somatic evolution, a Darwinian process that occurs with every infection. In the clonal selection model of immunogenesis (5), an apparently random production of immunoglobulin variants, both by reassortment of parts and by localized mutagenesis, gives rise to candidate antibodies, which then proliferate in response to matching epitopes. We do not understand the details of how a given epitope enhances stepwise improvements in affinity and productivity of antibodies at various stages. The process may be more complicated than we realize; so may Darwinian evolution.
Despite the prior arguments against relying on host or genotype evolution as a response to infection, historically we have done so and now have "scars of experience." A notable example is malaria, wherein the Duffy mutation against Plasmodium vivax is the only host defense with no deleterious consequences. The thalassemias, G6PD deficiency, and hemoglobin S are all hemopoietic modifications that thwart the plasmodia; but in homozygotes, they themselves cause disease. In the evolution of our species, for every child spared an early death because a hemoglobin S mutation impeded Plasmodium development, another will succumb to sickle cell disease unless we can intervene. Specific remedies do not exist. Although somatic gene therapy is an interesting possibility, one that will probably progress in the next 20 years, it is paradoxical that we know more about hemoglobin S than any other molecular disease. The entire concept of genetic determination of protein structure has been based on these early observations, yet we are still searching with limited success for ways to put it to therapeutic use.
Biotechnology may enable other forms of genetic intervention through which homo sapiens could conceivably bypass natural selection and random variation. In the absence of alternatives, we might speculate about these kinds of "aversive therapies" as a last resort to save our species.
The ultimate origin of life is still the subject of many theories, as is the origin of viruses (Table 2). Each virus is different. We know nothing of virus phylogenies and cannot even substantiate the distinctions of the several hundred categories. We do not know their origin, only that they interact with host genomes in many ways. Particles could come out of any genome, become free-living (i.e., independent, autonomously replicating units in host cells), reenter a host genome as retroviruses and possibly others do, and repeat the cycle dozens of times. But no one can give a single example or claim to have significant knowledge of how any particular virus evolved, thus presenting a scientific challenge for the next 20 or 30 years.
We are dealing with more than just predation and competition. We are dealing with a very complicated coevolutionary process, involving merger, union, bifurcation, and reemergence of new species (Table 3). Divergent phenomena can occur in any binary association, with unpredictable outcomes. We have hundreds of retroviruses in our genome and no knowledge of how they got there. As to HIV, we have no evidence as yet that it has ever entered anyone's germ line genome: we really do not know whether it ever enters germ cells. The outcomes of even that interaction could be much more complicated than the purely parasite/host relationships we are accustomed to.
Innovative technologies for dealing with microbial threats have the potential for fascinating therapeutic opportunities (Table 4). Some, like bacteriophage, have been set aside as laboratory curiosities. Nothing is more exciting than unraveling the details of pathogenesis. Having the full genomes of half a dozen parasitic organisms opens up new opportunities for therapeutic invention in ways that we could not have dreamed of even 5 years ago, which will lead to many more technologies. In food microbiology, we should keep in mind the probiotic as well as the adversarial and pathogenetic opportunities in our alimentary tracts.
The Committee on International Science Engineering and Technology report (2) provides some recommendations (Table 5). We need a global perspective. We need to invest in public health, especially food microbiology, not just medical care, in dealing with disease. It is important to prevent foodborne disease through sensible monitoring, standards of cleanliness, and consumer and food-handler education and not just care for its victims.
Today we emphasize individual rights over community needs more than we did 50 to 75 years ago. Restraining the rights and freedoms of individuals is a far greater sin than allowing the infection of others. The restraints placed on Typhoid Mary might not be acceptable today, when some would prefer to give her unlimited rein to infect others, with litigation their only recourse. In the triumph of individual rights, the public health perspective has had an uphill struggle in recent pandemics.
Education, however, is a universally accepted countermeasure, especially important in foodborne diseases. Food safety programs should more specifically target food handlers, examining their hands to determine if they are carriers, to ensure they are complying with basic sanitation.
We typically do this only after an outbreak. Perhaps we should have further debate on the social context for constraints and persuasion to contain the spread of infectious agents.
References
- Taubenberger JK, Reid AH, Frafft AE, Bijwaard KE, Fanning TG. Initial genetic characterization of the 1918 "Spanish" influenza virus. Science. 1997;275:1793–6. DOIPubMedGoogle Scholar
- Working NSTC-CISET. Group on Emerging and Reemerging Infectious Diseases. Infectious disease—a global health threat. Washington (DC): The Group;1996.
- Lederberg J. Plasmid (1952-1997). Plasmid. 1997. In press.
- Roelke-Parker ME, Munson L, Packer C, Kock R, Cleaveland S, Carpenter M, A canine distemper virus epidemic in Serengeti lions (Panthera leo). Nature. 1996;379:441–5. DOIPubMedGoogle Scholar
- Lederberg J. The ontogeny of the clonal selection theory of antibody formation: reflections on Darwin and Ehrlich. Ann N Y Acad Sci. 1988;546:175–87. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 3, Number 4—December 1997
| EID Search Options |
|---|
|
|
|
|
|
|