Volume 30, Number 6—June 2024
Dispatch
Autochthonous Plasmodium vivax Infections, Florida, USA, 2023
Cite This Article
Citation for Media
Abstract
During May–July 2023, a cluster of 7 patients at local hospitals in Florida, USA, received a diagnosis of Plasmodium vivax malaria. Whole-genome sequencing of the organism from 4 patients and phylogenetic analysis with worldwide representative P. vivax genomes indicated probable single parasite introduction from Central/South America.
Although commendable progress for combating malaria in endemic areas has been achieved and a dozen countries have been declared malaria-free since 2000 (1), increasing international travel has led to a rise in imported malaria cases, as well as sporadic cases of autochthonous malaria in non–malaria-endemic regions, especially those with competent vectors and favorable transmission conditions (2). In 2003 and 2023, two malaria outbreaks with local Plasmodium vivax transmission were reported in Florida (3,4). We examined the genomic characteristics, probable transmission dynamics, and likely origins of the 2023 P. vivax strains in Florida, demonstrating the role of genomic epidemiology in malaria control in non–malaria-endemic regions.
Figure 1
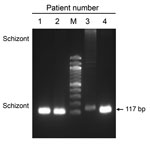
Figure 1. Identification of Plasmodium vivax infections in blood samples from malaria patients, Florida, USA, May–July 2023. Image shows 117-bp PCR products amplified from blood samples from 4 patients by using ...
During May–July 2023, the Florida Department of Health received reports of a series of 7 cases of P. vivax malaria (4). The patients lacked risk factors for contracting malaria (e.g., recent histories of international travel, blood transfusion, or previous malaria), raising concerns for local mosquito transmission. All 7 patients were concentrated within a 4-mile radius, raising further concern about potential local transmission cycles. Pretreatment blood samples from 4 patients were collected on June 18, 20, 27, and July 12 (Appendix Figure 1). We used the anonymized remnant clinical samples collected for diagnosis purposes. Species-specific PCR of the samples (5) confirmed P. vivax infections (Figure 1). Three Anopheles mosquitoes collected in the affected region in June also tested positive for P. vivax (4).
To trace the origin of those P. vivax cases, we isolated DNA from 200 μL of whole blood from 4 patients by using a QIAamp DNA Blood Mini Kit (QIAGEN, https://www.qiagen.com). Recognizing the limited P. vivax DNA contaminated with overwhelming amounts of human DNA, we performed selective whole-genome amplification (sWGA) with P. vivax–specific primers (pvset1920) to enrich the P. vivax genome (6). Each 50-µL amplification reaction included 30 U of phi29 polymerase enzyme (New England Biolabs, https://www.neb.com), 1% bovine serum albumin, 2 mM of each of the 4 deoxynucleosides triphosphate, 3.5 µM of selective whole-genome amplification primers, and ≈70 ng of input DNA. Thermocycler conditions consisted of ramping down from 35°C to 30°C at a rate of 0.1°C per minute, 30°C for 16 hours, and 65°C for 10 minutes. The reaction product was purified with AMPure XP beads (Beckman-Coulter, https://www.beckmancoulter.com) and eluted in 50 µL Tris-EDTA buffer (pH 8). We obtained 10–12 μg of amplification product for each sample.
The sequencing libraries were prepared by using an Illumina DNA Prep Kit (https://www.illumina.com) and were pooled and sequenced for 100 million reads at 150-bp paired-end reads mode on the Illumina Nextseq 2000 platform. After trimming adapters and removing low-quality reads (7), we aligned high-quality reads to the P. vivax PvP01 reference genome by using the BWA-MEM (Burroughs-Wheeler Aligner–maximum exact matches) algorithm (8). We compressed, sorted, and indexed the alignment files by using SAMtools version 1.11 (9). For the 4 samples, we mapped 3%, 16%, 29%, and 55% of total reads to the PvP01 genome (Table), resulting in 7–124-fold and ≈72% coverage of the PvP01 genome. We deposited sequence data in the National Center for Biotechnology Information BioProject database (https://www.ncbi.nlm.nih.gov; BioProject no. PRJNA1093439).
We performed variant calling by using the GATK HaplotypeCaller (https://gatk.broadinstitute.org). To retain only high-confidence variants, we applied stringent quality filtering criteria to the VCF (variant call format) files. Comparison with the PvP01 sequence identified 97,180 high-quality single-nucleotide polymorphisms (SNPs), of which 38,799 were shared among all 4 Florida P. vivax isolates.
Next, we calculated the within-host fixation index for each patient sample, which was >0.95, indicating monoclonal infections. Of note, 57,656 (≈60%) SNPs were shared among samples 2, 3, and 4, indicating their close genetic relationships. The level of genetic variation was similar to the limited variation found in meiosis of single crosses (10), suggesting a limited local transmission event, likely seeded by a single introduction into Florida.
Figure 2
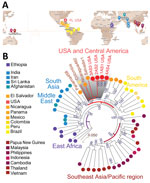
Figure 2. Phylogenetic analysis of Plasmodium vivaxstrains from blood samples from malaria patients, Florida, USA, May–July 2023, suggesting Central/South America origin. A) Geographic distribution of 53 high-quality global strains selected...
We constructed phylogeny between the Florida P. vivax isolates and 53 selected genomes from a global collection of 1,041 isolates (MalariaGen Pv4) (11). We chose a dataset consisting of high-quality P. vivax genome sequences from all continents where P. vivax is endemic (Figure 2, panel A). We constructed phylogenetic trees by using IQtree version 2.2.0 (12) and the general time-reversible model with ascertainment bias corrrection or the general time-reversible model with proportion of invariable sites and gamma-distributed rate heterogeneity. We evaluated both models with 1,000 ultrafast bootstrap approximation replicates and Shimodaira-Hasegawa–like approximate likelihood ratio test iterations. We assigned the final phylogenetic tree on the basis of bootstrap support values, ensuring robust statistical support for the inferred relationships. To ensure the reliability of the phylogenetic models, we allowed both models to converge; convergence was assessed at 300 iterations. We visualized and annotated the generated tree file by using Interactive Tree Of Life v.6 (13). Phylogenetic analysis with genome-wide SNPs revealed continental P. vivax population division (Figure 2, panel B) as previously reported (11). The P. vivax isolates from Florida, showing a pairwise genome distance of <0.01, formed a tight cluster (Appendix Figure 2), suggesting that they were probably the progeny of a single parasite isolate. Those isolates formed a highly supported Central/South America cluster (Figure 1). In sum, the timeline of the 7 cases (3,4) and our phylogenomic analysis support the interpretation of a single, limited introduction event from Central/South America into Florida.
Each year in the United States, ≈2,000 cases of imported malaria are reported to the Centers for Disease Control and Prevention (14). Although most cases are caused by P. falciparum infections, the 2003 and 2023 autochthonous outbreaks in Florida were caused by P. vivax (3,4). Similarly, local outbreaks from introduced P. vivax in Greece (2) and the Republic of Korea (15) illustrate the high potential for imported P. vivax to establish sustained transmission, which may pose a substantial challenge for subsequent elimination. Although the risk for autochthonous malaria in the United States remains low, the potential threat of imported P. vivax setting off and establishing local transmission in areas with competent vectors and conducive environments is a public health concern.
The last mosquito-vectored P. vivax malaria outbreak in the United States was in 2003 in Palm Beach County, Florida, and involved a cluster of 7 cases (3) before the P. vivax genome sequence was available. Although microsatellites were used to identify their genetic relatedness, the genomic tools that we used offer unparalleled resolution of the genetic relatedness of parasite isolates of the 2023 malaria outbreak in Florida. The minimal genetic variations among the isolates indicate a single transmission chain. Phylogenetic clustering with P. vivax strains from Central/South America implies the travel-related import of the index isolate from that region. Of note, a case of imported P. vivax malaria with symptom onset April 20, 2023, was reported from the same area as the 7 P. vivax cases (3,4). We are unable to determine if the April case initiated local transmission because no remaining specimen was available for analysis.
In summary, our study underscores the usefulness and power of genomic tools in epidemiologic investigations. The established analytical pipeline will enable more streamlined and efficient genomic surveillance, provide health authorities with accurate information about the source of the infections, and enable communication of the risk to the public with solid scientific evidence. Given the rich genomic resources of worldwide P. vivax isolates, genomic surveillance will play a crucial role in tracking additional cases, detecting potential transmission chains, and identifying relapse. Although locally transmitted malaria in Florida has been successfully eliminated, ongoing surveillance, rapid responses, vigilance, and preparedness are still needed to prevent malaria reintroduction and local transmission.
Dr. Muneer is a postdoctoral scholar at the University of South Florida. His current focus is related to drug resistance in malaria parasites.
Acknowledgments
We acknowledge the contribution of Min Zhang, the University of South Florida Genomics Program, and the USF Omics Hub for completion of this project. We thank Brian Raphael and Joel Barratt for helpful discussions and critical review of the manuscript.
The study is supported by grant no. U19AI089672 from the National Institute of Allergy and Infectious Diseases, National Insitutes of Health.
References
- World Health Organization. World Malaria Report 2023. Geneva, Switzerland: The Organization; 2023.
- Spanakos G, Snounou G, Pervanidou D, Alifrangis M, Rosanas-Urgell A, Baka A, et al.; MALWEST Project. MALWEST Project. Genetic spatiotemporal anatomy of Plasmodium vivax malaria episodes in Greece, 2009–2013. Emerg Infect Dis. 2018;24:541–8. DOIPubMedGoogle Scholar
- Centers for Disease Control and Prevention (CDC). Local transmission of Plasmodium vivax malaria—Palm Beach County, Florida, 2003. MMWR Morb Mortal Wkly Rep. 2003;52:908–11.PubMedGoogle Scholar
- Blackburn D, Drennon M, Broussard K, Morrison AM, Stanek D, Sarney E, et al. Outbreak of locally acquired mosquito-transmitted (autochthonous) malaria—Florida and Texas, May–July 2023. MMWR Morb Mortal Wkly Rep. 2023;72:973–8. DOIPubMedGoogle Scholar
- Snounou G, Viriyakosol S, Zhu XP, Jarra W, Pinheiro L, do Rosario VE, et al. High sensitivity of detection of human malaria parasites by the use of nested polymerase chain reaction. Mol Biochem Parasitol. 1993;61:315–20. DOIPubMedGoogle Scholar
- Cowell AN, Loy DE, Sundararaman SA, Valdivia H, Fisch K, Lescano AG, et al. Selective whole-genome amplification is a robust method that enables scalable whole-genome sequencing of Plasmodium vivax from unprocessed clinical samples. MBio. 2017;8:e02257–16. DOIPubMedGoogle Scholar
- Andrews S. FastQC: a quality control tool for high throughput sequence data. Cambridge (UK): Babraham Bioinformatics, Babraham Institute; 2010.
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. DOIPubMedGoogle Scholar
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. DOIPubMedGoogle Scholar
- Bright AT, Manary MJ, Tewhey R, Arango EM, Wang T, Schork NJ, et al. A high resolution case study of a patient with recurrent Plasmodium vivax infections shows that relapses were caused by meiotic siblings. PLoS Negl Trop Dis. 2014;8:
e2882 . DOIPubMedGoogle Scholar - Adam I, Alam MS, Alemu S, Amaratunga C, Amato R, Andrianaranjaka V, et al.; MalariaGEN. An open dataset of Plasmodium vivax genome variation in 1,895 worldwide samples. Wellcome Open Res. 2022;7:136. DOIPubMedGoogle Scholar
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587–9. DOIPubMedGoogle Scholar
- Letunic I, Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49(W1):W293–6. DOIPubMedGoogle Scholar
- Mace KE, Lucchi NW, Tan KR. Malaria Surveillance - United States, 2018. MMWR Surveill Summ. 2022;71:1–35. DOIPubMedGoogle Scholar
- Bahk YY, Lee HW, Na BK, Kim J, Jin K, Hong YS, et al. Epidemiological characteristics of re-emerging vivax malaria in the Republic of Korea (1993–2017). Korean J Parasitol. 2018;56:531–43. DOIPubMedGoogle Scholar
Figures
Table
Cite This ArticleOriginal Publication Date: April 25, 2024
1These authors contributed equally to this article.
2These authors were co-principal investigators.
Table of Contents – Volume 30, Number 6—June 2024
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Liwang Cui, Division of Infectious Disease and International Medicine, Department of Internal Medicine, Morsani College of Medicine, University of South Florida, 3720 Spectrum Blvd, Ste 304, Tampa, FL 33612, USA
Top