Volume 30, Number 8—August 2024
Research
Phylogeographic Analysis of Mycobacterium kansasii Isolates from Patients with M. kansasii Lung Disease in Industrialized City, Taiwan
Cite This Article
Citation for Media
Abstract
Little is known about environmental transmission of Mycobacterium kansasii. We retrospectively investigated potential environmental acquisition, primarily water sources, of M. kansasii among 216 patients with pulmonary disease from an industrial city in Taiwan during 2015–2017. We analyzed sputum mycobacterial cultures using whole-genome sequencing and used hierarchical Bayesian spatial network methods to evaluate risk factors for genetic relatedness of M. kansasii strains. The mean age of participants was 67 years; 24.1% had previously had tuberculosis. We found that persons from districts served by 2 water purification plants were at higher risk of being infected with genetically related M. kansasii isolates. The adjusted odds ratios were 1.81 (1.25–2.60) for the Weng Park plant and 1.39 (1.12–1.71) for the Fongshan plant. Those findings unveiled the association between water purification plants and M. kansasii pulmonary disease, highlighting the need for further environmental investigations to evaluate the risk for M. kansasii transmission.
Mycobacterium kansasii, a slow-growing nontuberculous mycobacteria (NTM), can cause destructive pulmonary diseases in humans that cause similar clinical manifestations to those of M. tuberculosis pulmonary disease (1). In recent years, M. kansasii has become one of the most frequently reported NTMs in the world (2), but whether increasing notifications in some countries (3) reflects true increases in the incidence of M. kansasii disease or improvements in laboratory identification is not well understood.
Infection with M. kansasii has been predominantly reported in urban settings, in high-density and low-income communities, and among gold miners (4). Despite recent reports of potential human-to-human transmission for other NTMs (5), M. kansasii lung disease has been generally assumed to be acquired from environmental sources (6). Nonetheless, the precise route of transmission is yet to be characterized. M. kansasii is ubiquitous in the environment; tap water is reported as a major reservoir (6). The waterborne acquisition of M. kansasii is enabled by its intrinsic resistance to disinfectants, acid, and heat (7); its ability to survive in oligotrophic water; and its ability to form pipe surface biofilms (8). In previous studies, M. kansasii has been isolated from the water distribution system in the same communities in which cases with M. kansasii disease arise (9). However, evidence on environmental acquisition on the basis of genotyping of clinical or environmental isolates has been limited.
We investigated the environmental acquisition of M. kansasii in the industrial city of Kaohsiung, located in southern Taiwan. Our earlier multicenter study revealed that the number of patients with M. kansasii pulmonary infection was nearly 5-fold higher in Kaohsiung than in Taipei in northern Taiwan (10). Higher humidity levels, warmer temperatures, and industrial areas with greater air pollution in Kaohsiung might explain this difference. We previously identified 2 spatial hotspots of high risk for M. kansasii infection in Kaohsiung by using clinical data collected from patients in all tertiary medical centers of the city (11). We hypothesized that specific water supplies or heavy industrial areas might be associated with the risk for M. kansasii infection. In this study, we extended the previous spatial analysis to include whole-genome sequencing (WGS) data of clinical M. kansasii isolates along with geographic information from patients, area-level industrialization, and water supply systems to comprehensively assess the risk and drivers of M. kansasii in Kaohsiung.
Study Participants
We included 302 patients >20 years of age who had newly diagnosed M. kansasii lung disease, consistent with American Thoracic Society/Infectious Diseases Society of America guidelines (12) from 1 tertiary medical center and its affiliated regional hospitals in Kaohsiung during 2015–2017. We conducted chart reviews to collect clinical and demographic data from eligible persons.
Mycobacterial WGS
Of the 302 patients, we performed WGS on mycobacterial isolates from 243 patients with high-quality culture samples from pretreatment sputum or bronchoalveolar lavage fluid. We performed sequencing in the Laboratory of Genomics and Bioinformatics Service at Texas A&M AgriLife (College Station, TX, USA) and constructed libraries by using the NEXTFLEX Rapid XP DNA-Seq Kit (Revvity, https://www.revvity.com). We performed paired-end sequencing using the NovaSeq 6000 Sequencing System (Illumina, https://www.illumina.com) with a read length of 150 bp and anticipated minimum mean depth of coverage of 100×. We removed 22 samples with most reads from a species other than M. kansasii from analysis. One additional sample had a large degree of nonmycobacterial reads, and 4 samples were from participants missing clinical or residential data. We excluded those from analysis, leaving 216 samples for our primary analysis.
Environmental Exposure Data
Figure 1
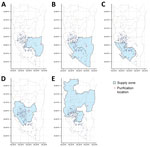
Figure 1. Spatial distribution of residential addresses of Mycobacterium kansasii isolates from patients, water purification plants, and corresponding supply zones in phylogeographic analysis of M. kansasiiisolates from patients...
Our primary environmental exposure of interest was the water sources. We collected information on the service areas of water purification plants in Kaohsiung through the official website of Taiwan Water Corporation (13). In total, 5 water purification plants provide service to most households in Kaohsiung. Among those, Pingding is the largest by volume (44% of the total water supply from the 5 plants), followed by Chengcing (29%), Kaotan (12%), Fongshan (10%), and Weng Park (4%) (13). The water supply networks of those purification plants overlap; a single district often receives service from >1 water plant (Figure 1). We identified areas of heavy industrial zoning in Kaohsiung and used a probabilistic approach to identify participants who had a high probability of working in 1 of those zones (Appendix).
Data Analysis
We classified and filtered raw fastq reads using kraken2 (14) with a custom database, bracken (15), and ntm-profiler (16) (Appendix). We used Shovill (17) for de novo assembly using SPAdes (18). We aligned assemblies using ska (S.R. Harris, unpub. data, https://www.biorxiv.org/content/10.1101/453142v1) to the M. kansasii strain ATCC 12478, and all had >90% genomic coverage of the reference. We identified and masked regions of horizontal sequence transfer using Gubbins (19) and constructed a recombination-masked pairwise single-nucleotide polymorphism (SNP) distance matrix with snp-dists (20). We used this structure to create a phylogenetic tree with RAxML-NG (21) using the M. kansasii strain FDA-ARGOS 1615 as an outgroup, a generalized time reversible with gamma model of rate heterogeneity, and 50 starting trees (25 random and 25 parsimony) and calculated the transfer bootstrap expectation metric. To assess the association between genetic clade and water purification plant, we used Pearson χ2 tests.
To assess the sensitivity of our phylogenetic analysis, we repeated the analysis with the addition of genomic sequences obtained from public sources and identified as obtained from outside of Taiwan. We used a list of available isolates (9) to download short-read sequences of 22 specimens from the National Center for Biotechnology Information Sequence Read Archive database. We speciated and filtered the sequences with the same analysis pipeline as our samples from Taiwan and repeated phylogenetic analyses using a tree constructed from the combined sample sets.
To analyze factors affecting the genetic relatedness of M. kansasii strains between pairs of persons from the study sample, we used hierarchical Bayesian spatial network methods implemented in the R package GenePair (22). As opposed to standard regression modeling, GenePair methods are designed to account for correlation observed in dyadic data given that the same person is represented across multiple pairs (i.e., network dependence), spatial correlation between paired outcomes because of unmeasured transmission dynamics, and specific distributional features of genetic relatedness outcomes (23).
We modeled 2 outcomes of genetic relatedness: a binary outcome in which we classified pairs of strains as clustered or not based on a threshold SNP distance and a continuous measure of SNP distance between pairs of strains. For the binary outcome, the bimodal distribution of observed SNP distances between strains (Appendix Figure 1) suggested a threshold of <45 SNPs to classify pairs of strains as being within a cluster. In a sensitivity analysis, we also investigated a more conservative cutoff of <32 SNPs. We fit all models using Markov chain Monte Carlo sampling techniques and assessed convergence using trace plots and Geweke’s diagnostic for all relevant model parameters. When making statistical inference, we reported posterior means and 95% equal-tailed quantile-based credible intervals.
Independent variables included the time between the date of each participant’s sputum collection in days, the combined age of both participants in years, the age difference between pairs of participants in years, whether both were male or female, whether either person in the pair had cavitary disease on chest radiograph, geographic distance between participant households in kilometers, whether participant households were both supplied by the same water purification plants, whether both participants resided in villages with a high degree of linkage to the 4 heavy industrial zones, and whether the patients had received their diagnosis and been treated in the same hospital. We chose demographic and clinical variables included in the models a priori on the basis of clinical relevance.
The study was approved by the institutional ethics committees of the participating hospitals (KMUHIRB-F(I)-20210173, KMUHIRB-E(I)-20210380). The study funders had no role in the design and conduct of the study, the analysis and interpretation of data, or in the preparation, review, or approval of the manuscript.
Demographics
Of the 216 cases with complete information, the mean patient age was 67 years (SD 17.6); 146 (67.6%) were men and 70 (32.4%) women (Table). Mean body mass index was 21 (SD 4.1). Of the 216 case-patients, 46 (21.3%) were current smokers, 45 (20.8%) were ex-smokers, and 125 (57.9%) had never smoked. On chest radiograph, 42 (20.1%) persons had consolidation, 37 (17.1%) had fibrocavitary disease, 81 (38.8%) had nodular bronchiectasis, 3 (1.4%) had nodules, 1 (0.5%) had fibrosis, and 52 (24.1%) had mixed image patterns. Tuberculosis had been diagnosed previously in 52 (24.1%) persons.
Sources of Residential Water Purification
Participant households were served by a median of 2 (maximum 3) of 8 different water purification plants. Chengcinghu supplied 128 (59.3%) of participant households, Pingding supplied 84 (38.9%), Kaotan supplied 99 (45.8%), Fongshan supplied 70 (32.4%), Weng Park supplied 22 (10.2%), Lingkou supplied 4 (1.9%), and Baolai and Lujhu each supplied 1 (0.5%) (Table; Figure 1).
Factors Associated with Genetic Relatedness between Pairs of Participants
Figure 2
![Associations of environmental and clinical risk factors with genetic relatedness based on pair-level data using hierarchical Bayesian regression models in phylogeographic analysis of Mycobacterium kansasii isolates from patients with M. kansasii lung disease in industrialized city, Taiwan. A) Odds ratios for pairs of M. kansasii isolates to be in a genetic cluster (using the single-nucleotide polymorphism [SNP] cutoff of 45). An odds ratio of >1 suggests that the risk factor was associated with genetic clustering. B) Risk ratios for increase in SNP distance between pairs of isolates. A risk ratio of <1 suggests that the risk factor was associated with a shorter pairwise SNP distance. The 3 smaller water purification plants (Lingkou, Baolai, and Lujhu) were not considered in the analysis as they together only provided service to 6 participants.](/eid/images/24-0021-F2-tn.jpg)
Figure 2. Associations of environmental and clinical risk factors with genetic relatedness based on pair-level data using hierarchical Bayesian regression models in phylogeographic analysis of Mycobacterium kansasii isolates from patients with ...
Before analyzing genetic relatedness of M. kansasii strains, we masked areas of horizontal sequence transfer as previously described. This process reduced the median number of SNPs per strain from 23 (interquartile range [IQR] 537) to 13 (IQR 14.25). We constructed hierarchical Bayesian models to evaluate factors associated with being part of a genetic cluster (i.e., M. kansasii SNP distance between isolates of <45) as well as factors associated with continuous SNP distance. The model results (Figure 2) indicated that for each pair of participants, if both participant residences were supplied by the Weng Park water purification plant, their M. kansasii isolates had increased likelihood of being genetically clustered (odds ratio [OR] 1.81, 95% credible interval [CrI] 1.25–2.60), after adjusting for the other relevant factors. This finding was mirrored in the SNP model, where the SNP distances between the M. kansasii isolates of those pairs of persons were ≈12% smaller on average (risk ratio [RR] 0.88, 95% CrI 0.85–0.92). With a clustered model and a more conservative cutoff of 32 SNPs, the magnitude of the association was similar but not statistically significant (OR 1.54, 95% CrI 0.96–2.45) (Appendix Figure 2). We found a lower magnitude of association for the Fongshan water purification plant with a clustering OR of 1.39 (95% CrI 1.12−1.71) and a RR of SNP distance of 0.96 (0.94–0.98). We observed an inverse but statistically insignificant association between linear spatial distance and odds of clustering; adjusted OR was 0.77 (95% CrI 0.20–2.88) for every 1 km increase in spatial distance. Having a linkage to the same heavy industrial zone and sharing the same healthcare facility were not significantly associated with genetic relatedness, nor did they significantly alter the effect sizes for other variables in the model. Therefore, we removed those 2 variables from the final multivariable models for parsimony.
The statistical models also provided an estimate of a spatially referenced random effect parameter for each person that described the participants’ residual (after adjustment for other factors) risk of being infected with an M. kansasii isolate that was genetically similar to other participants in the study. We mapped posterior mean estimates for those parameters (Appendix Figure 3). From visual inspection, we noted no obvious areas of increased residual risk.
Tree Structure of M. kansasii Population
Figure 3

Figure 3. Maximum-likelihood phylogram of clinical Mycobacterium kansasii isolates from patients with M. kansasiilung disease in industrialized city, Taiwan. Phylogeny with major clades are labeled as A, B,...
We created a maximum-likelihood phylogram that showed 3 main clades (Figure 3). Participants with residential water supplied by the Weng Park water purification plant were more likely to be in clade A. Of the 73 persons in clade A, 14 (19.2%) were supplied by Weng Park. Of 58 participants in clade B, 3 (5.2%) had water purified by Weng Park; 5 (6.1%) of 82 participants in clade C had water purified by Weng Park (p = 0.008 for the association between genetic clade and Weng Park). Participants supplied by the Fongshan water purification plant were more likely to be in clade B. Of the 58 participants in clade B, 27 (46.6%) were supplied by Fongshan. Of 73 participants in clade A, 15 (20.5%) were supplied by Fongshan; 27 (32.9%) of 82 in clade C were supplied by Fongshan (p = 0.006 for the association between genetic clade and Fongshan). We conducted a sensitivity analysis that included an additional 22 publicly available M. kansasii isolates from outside Taiwan in the phylogenetic analysis. The resulting phylogeny revealed the 3 major clades, which contained the same Taiwan samples as in the main analysis (Appendix Figure 4). Similarly, when we constructed a phylogeny of our samples from Taiwan without masking recombinant regions, the same clades were identified with identical members in clades A and B. Clade C in the phylogeny constructed without recombination masking contained all of the samples seen in the primary analysis but also overlapped with 59 samples from clade B.
Using densely sampled cases of M. kansasii infection from a tropical metropolitan city in southern Taiwan, we investigated environmental factors associated with genetic relatedness using WGS data. We found that pairs of persons with M. kansasii lung disease living in a district served by specific water purification plants (Weng Park and Fongshan) were at greater risk of being infected with genetically similar M. kansasii isolates. The association of smaller spatial distances between pairs of persons had a statistically significant trend toward more genetically similar M. kansasii strains.
We previously conducted a spatial analysis of 537 M. kansasii cases from 4 major tertiary hospitals in Kaohsiung and identified 2 suspicious spatial clusters (11). In this analysis, we found that the Weng Park and Fongshan water purification plants, both near 1 of the previously identified spatial hotspots, were associated with increased genetic relatedness among case pairs.
Weng Park, the water purification plant with the strongest signal of genetic relatedness (adjusted OR 1.81, 95% CrI 1.25–2.60) among M. kansasii cases, accounted for only 4% of the total water supply in the study area but was associated with 22 (10%) of the total 216 cases. We also observed that Weng Park was significantly associated with 1 major M. kansasii clade in our phylogenic analysis. Because water from different water plants mixed in underground water pipes, determining the source of supply for a particular household was difficult. Given the relatively low number of households supplied by Weng Park, the cases and households labeled as Weng Park might have also received their supply from other water plants, such as Kaotan and Pingding (Figure 1). The misclassification of water purification plants at the household level would lead us to underestimate the association between Weng Park and genetic relatedness of M. kansasii among case pairs.
This study does not provide a mechanism for the association between Weng Park and Fongshan water plants and the genetic clustering of M. kansasii–infected cases. Each water plant applied different methods of water purification, resulting in differences in pH value or organic matter content that might affect the risk for contamination and growth of M. kansasii (6). Different sources of raw water might also potentially influence the microbiological ecology; a 2003 analysis found that water from Weng Park had substantially higher general hardness than other water plants, suggesting a higher contribution from underground water as the source (13). The presence of more sediment accumulation in pipelines from underground water than from surface water might accelerate biofilm development (24). As an example of the effect of the water treatment and distribution system on NTM abundance, a previous study in the United States reported low NTM relative abundances in Mississippi River water, a source for the drinking water system, but high relative abundances in the distribution system and tap water (25). Further environmental samplings should be conducted to examine the distribution of M. kansasii isolates in different water sources and plants, particularly Weng Park and Fongshan water plants.
Recent analyses of M. abscessus, another pathogenic NTM, have suggested that transmission might occur from person-to-person, especially in cystic fibrosis patients who might attend the same clinic (5). Our study did not find a potential link between shared clinic visits and the genetic clustering of M. kansasii in the investigated cases by analyzing pairs of persons’ strains treated at the same hospital.
Although WGS has been widely applied to understand the transmission dynamics of M. tuberculosis, its application for studying NTM transmission including M. kansasii is still limited (9,26,27). The population structure and genomic diversity of M. kansasii on a global scale have been previously reported (28), but the genomic diversity over a well-defined geography has not been characterized. We used the cutoff of 45 and 32 SNPs to define genetic relatedness on the basis of the empirical SNP distribution in our population, but the relationship between those measures of genetic distance and transmission remains uncertain. A major difference between M. tuberculosis and M. kansasii is that genomic recombination occurs frequently in M. kansasii through distributive conjugal transfer (28). A recent study using 60 samples of M. kansasii from different provinces of China revealed that the pairwise SNP distance (after masking recombinant regions) of those isolates were all within 20 SNPs and suggested a threshold of 4 SNPs to define clustering (29). Both that study and ours excluded recombinant regions using the same methods, further complicating the interpretation of our analysis in terms of transmission inference. In addition, selecting an effective threshold to cluster cases with a potential shared exposure site might be dependent on the local epidemiology and genetic diversity of the tested population. Along with this study, 2 other large analyses of clinical M. kansasii whole-genome sequences (9,29) showed that, after masking recombinant regions, the resulting sequences appeared to belong to homogeneous clusters with maximum SNP distances on the order of 100 SNPs. The largest analysis to date created a phylogeny that estimated the most recent common ancestor of a global collection of clinical isolates to be timed to the early 1900s (9).
A major strength of this study is the combination of WGS and detailed spatial information on the environmental determinants of interest, including the water supply and heavy industrial zoning. The novel Bayesian hierarchical modeling approach correctly accounts for the correlation of pairwise spatial-genetic data and the simultaneous adjustment for potential confounders. Previous studies on the transmission of M. kansasii have mostly applied conventional genotyping methodologies (e.g., restriction fragment length polymorphism or targeted PCR analysis) (30,31).
The first limitation of this study is that the M. kansasii isolates came from 1 major tertiary medical center and its affiliated hospitals (accounting for 56% of all M. kansasii isolates from all major medical centers in Kaohsiung during the same period) (11). M. kansasii infection is not a notifiable disease in Kaohsiung, and thus the population coverage of this analysis is not comprehensive. The suboptimal population coverage posed a challenge in identifying environmental exposures. Second, our analysis revealed an association between certain water purification plants (Weng Park and Fongshan) and genetic relatedness, but the route of transmission cannot be confirmed without environmental sampling. Weng Park only accounted for 10% of total cases and Fongshan accounted for 32% of cases. Environmental determinants of most M. kansasii cases remain to be elucidated. Third, we only have crude (district-level) spatial coverage of the water purification plants, and misclassification of water supply at the household level might well occur. Last, we used the residential address as the proxy for exposure assessment, and we were not able to obtain definitive workplace exposures.
In conclusion, our novel spatial phylogenetic analysis in a densely sampled, well-defined geography revealed an independent association between certain water purification plants and the genetic relatedness of M. kansasii isolates. Our approach demonstrated the utility of combining WGS sequencing and readily available clinical and environmental information to obtain useful insights into the transmission of M. kansasii to trigger further environmental investigation and control measures.
Dr. Cudahy is an infectious disease specialist and Assistant Professor at the Yale School of Medicine. His primary research interest is mycobacterial infection outcomes and epidemiology.
Acknowledgments
We thank laboratory members Mo-Hua Li and Yu-Wei Chen for helping with mycobacterial culture and DNA extraction of M. kansasii isolates.
Funding for this study was provided by Taiwan Ministry of Science and Technology (MOST 111-2628-B-002-045, MOST 112-2314-B-037 −073 -MY3), Kaohsiung Municipal Ta-Tung Hospital (KMTTH-111-039, KMTTH-112-R002), and National Health Research Institutes (NHRI-EX108-10805PI). P.G.T.C. was funded by a career development grant from the Fogarty International Center of the National Institutes of Health (5K01TW011194).
All genomes are shared through the US National Center for Biotechnology Information Sequence Read Archive.
P.G.T.C., P.C.L., H.L.H., T.C., and H.H.L. conceptualized the study. P.G.T.C., J.L.W., and T.C. did the genomic and statistical analyses. B.S., C.Y., and T.I. contributed to the genomic analyses. P.C.L. contributed to the spatial analysis. P.L.L. provided isolates for analyses. P.G.T.C., P.C.L., H.L.H., T.C., and H.H.L. wrote the initial draft of the manuscript. All authors edited and reviewed the final manuscript. All authors had full access to all the data in the study and had final responsibility for the decision to submit for publication.
References
- Huang HL, Lu PL, Lee CH, Chong IW. Treatment of pulmonary disease caused by Mycobacterium kansasii. J Formos Med Assoc. 2020;119(Suppl 1):S51–7. DOIPubMedGoogle Scholar
- Martín-Casabona N, Bahrmand AR, Bennedsen J, Thomsen VO, Curcio M, Fauville-Dufaux M, et al.; Spanish Group for Non-Tuberculosis Mycobacteria. Non-tuberculous mycobacteria: patterns of isolation. A multi-country retrospective survey. Int J Tuberc Lung Dis. 2004;8:1186–93.PubMedGoogle Scholar
- Marras TK, Chedore P, Ying AM, Jamieson F. Isolation prevalence of pulmonary non-tuberculous mycobacteria in Ontario, 1997 2003. Thorax. 2007;62:661–6. DOIPubMedGoogle Scholar
- Bolden N, Mell JC, Logan JB, Planet PJ. Phylogenomics of nontuberculous mycobacteria respiratory infections in people with cystic fibrosis. Paediatr Respir Rev. 2023;46:63–70. DOIGoogle Scholar
- Vaerewijck MJM, Huys G, Palomino JC, Swings J, Portaels F. Mycobacteria in drinking water distribution systems: ecology and significance for human health. FEMS Microbiol Rev. 2005;29:911–34. DOIPubMedGoogle Scholar
- Blanc SM, Robinson D, Fahrenfeld NL. Potential for nontuberculous mycobacteria proliferation in natural and engineered water systems due to climate change: a literature review. City Environ Interact. 2021;11:
100070 . DOIGoogle Scholar - Gan Y, Rahmatika I, Kurisu F, et al. The fate and risk of nontuberculous mycobacteria in the water supply system: a review. H2Open J. 2022;5:180–197.
- Luo T, Xu P, Zhang Y, Porter JL, Ghanem M, Liu Q, et al. Population genomics provides insights into the evolution and adaptation to humans of the waterborne pathogen Mycobacterium kansasii. Nat Commun. 2021;12:2491. DOIPubMedGoogle Scholar
- Huang HL, Cheng MH, Lu PL, Shu CC, Wang JY, Wang JT, et al. Epidemiology and predictors of NTM pulmonary infection in Taiwan—a retrospective, five-year multicenter study. Sci Rep. 2017;7:16300. DOIPubMedGoogle Scholar
- Liu BC, Huang HL, Chan TC, Lee SJ, Lin JN, Lee CH, et al. Spatial cluster analysis of Mycobacterium kansasii infection in Kaohsiung, Taiwan. Taiwan J Public Health. 2021;40:713–23.
- Daley CL, Iaccarino JM, Lange C, Cambau E, Wallace RJ Jr, Andrejak C, et al. Treatment of nontuberculous mycobacterial pulmonary disease: an official ATS/ERS/ESCMID/IDSA clinical practice guideline. Clin Infect Dis. 2020;71:e1–36. DOIPubMedGoogle Scholar
- Kuo J, Chen Y. Statistical analysis and exploration of drinking water quality in Kaohsiung City. Kaohsiung City Government; 2007 [cited 2022 Sep 16]. https://ksepb.kcg.gov.tw/FileDownLoad/FileUpload/20191027122705617901.odt
- Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019;20:257. DOIPubMedGoogle Scholar
- Lu J, Breitwieser FP, Thielen P, Salzberg SL. Bracken: estimating species abundance in metagenomics data. PeerJ Comput Sci. 2017;3:
e104 . DOIGoogle Scholar - Prjibelski A, Antipov D, Meleshko D, Lapidus A, Korobeynikov A. Using SPAdes de novo assembler. Curr Protoc Bioinformatics. 2020;70:
e102 . DOIPubMedGoogle Scholar - Croucher NJ, Page AJ, Connor TR, Delaney AJ, Keane JA, Bentley SD, et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2015;43:e15–15. DOIPubMedGoogle Scholar
- Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics. 2019;35:4453–5. DOIPubMedGoogle Scholar
- Warren JL, Chitwood MH, Sobkowiak B, Colijn C, Cohen T. Spatial modeling of Mycobacterium tuberculosis transmission with dyadic genetic relatedness data. Biometrics. 2023;79:3650–63. DOIPubMedGoogle Scholar
- Hoff PD. Random effect models for network data. In: Dynamic social network modeling and analysis: workshop summary and papers. Washington: The National Academies Press; 2003. p. 303–12.
- Learbuch KLG, Smidt H, van der Wielen PWJJ. Water and biofilm in drinking water distribution systems in the Netherlands. Sci Total Environ. 2022;831:
154940 . DOIPubMedGoogle Scholar - Hull NM, Holinger EP, Ross KA, Robertson CE, Harris JK, Stevens MJ, et al. Longitudinal and source-to-tap New Orleans, LA, U.S.A. drinking water microbiology. Environ Sci Technol. 2017;51:4220–9. DOIPubMedGoogle Scholar
- Bryant JM, Grogono DM, Rodriguez-Rincon D, Everall I, Brown KP, Moreno P, et al. Emergence and spread of a human-transmissible multidrug-resistant nontuberculous mycobacterium. Science. 2016;354:751–7. DOIPubMedGoogle Scholar
- Nikolayevskyy V, Niemann S, Anthony R, van Soolingen D, Tagliani E, Ködmön C, et al. Role and value of whole genome sequencing in studying tuberculosis transmission. Clin Microbiol Infect. 2019;25:1377–82. DOIPubMedGoogle Scholar
- Tagini F, Pillonel T, Bertelli C, Jaton K, Greub G. Pathogenic determinants of the Mycobacterium kansasii complex: an unsuspected role for distributive conjugal transfer. Microorganisms. 2021;9:348. DOIPubMedGoogle Scholar
- Guo Y, Cao Y, Liu H, Yang J, Wang W, Wang B, et al. Clinical and microbiological characteristics of Mycobacterium kansasii pulmonary infections in China. Microbiol Spectr. 2022;10:
e0147521 . DOIPubMedGoogle Scholar - Picardeau M, Prod’Hom G, Raskine L, LePennec MP, Vincent V. Genotypic characterization of five subspecies of Mycobacterium kansasii. J Clin Microbiol. 1997;35:25–32. DOIPubMedGoogle Scholar
- Kwenda G, Churchyard GJ, Thorrold C, Heron I, Stevenson K, Duse AG, et al. Molecular characterisation of clinical and environmental isolates of Mycobacterium kansasii isolates from South African gold mines. J Water Health. 2015;13:190–202. DOIPubMedGoogle Scholar
Figures
Table
Cite This ArticleOriginal Publication Date: July 10, 2024
Table of Contents – Volume 30, Number 8—August 2024
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Hsien-Ho Lin, Institute of Epidemiology and Preventive Medicine, College of Public Health, National Taiwan University, Taipei 100, Taiwan
Top