Volume 31, Number 12—December 2025
Dispatch
Novel Highly Pathogenic Avian Influenza A(H5N1) Virus, Argentina, 2025
Cite This Article
Citation for Media
Abstract
Genomic sequencing of reemerging highly pathogenic avian influenza A(H5N1) virus detected in Argentina in February 2025 revealed novel triple-reassortant viruses containing gene segments from Eurasian H5N1 and low pathogenicity viruses from South and North American lineages. Our findings highlight continued evolution and diversification of clade 2.3.4.4b H5N1 in the Americas.
Highly pathogenic avian influenza (HPAI) viruses were introduced to South America in 2022 by migratory birds from North America. The viruses belonged to the 2.3.4.4b clade of HPAI A(H5N1) virus that became widespread in Europe in 2020 and spread to North America in 2021. The trajectory of H5N1 in South America has differed from H5N1 in North America in several critical ways. First, nearly all South America outbreaks stem from a single introduction of H5N1 viruses from North America (1,2), whereas the North America epizootic was reseeded by multiple independent introductions from Europe and Asia (A1–A6) (3,4). Second, South America H5N1 outbreaks were driven by a single genotype (B3.2) that was introduced from North America and remained genetically stable during its spread across South America. In contrast, H5N1 viruses in North America underwent frequent reassortment with low pathogenicity avian influenza (LPAI) viruses, prompting new genotype nomenclature (using B, C, D) (3). Third, South America’s H5N1 epizootic is unique in establishing mammal-to-mammal transmission in marine mammals, enabled by the H5N1 (B3.2) virus acquiring mammalian-adaptive polymerase basic (PB) 2 mutations (Q591K and D701N) (1,2). That pattern has not occurred in North America, where H5N1 spillover into terrestrial and marine mammals was transient, except in United States dairy cattle (3).
Beyond the ecologic devastation among coastal wildlife, in 2023, H5N1 (B3.2) virus spread widely in birds across mainland South America, leading to poultry and wild bird outbreaks (5–8). Although in 2024 HPAI outbreaks occurred in Brazil and Peru (World Organisation for Animal Health, https://wahis.woah.org), there were no detections in Argentina during March 2024–January 2025.
On February 11, 2025, Servicio Nacional de Sanidad y Calidad Agroalimentaria (SENASA; Buenos Aires, Argentina), Argentina’s national organization for agricultural health and safety, was notified of an outbreak in a mixed backyard flock (chickens, ducks, and turkeys) in Chaco Province, northern Argentina. The flock experienced high mortality (33/81 chickens, 37/99 ducks) in just 1 week. When we inspected the living flock, two thirds of the remaining 48 chickens had diarrhea and 1 of the remaining 62 ducks was lethargic; 2 turkeys were asymptomatic. The household was located within a remnant fragment of the Dry Chaco biome, a hot and semi-arid tropical dry forest, surrounded by agriculture cropland. The affected flock had free access to a small pond frequently visited by wild waterfowl (Appendix 1). We depopulated and disinfected the area. We inspected backyard poultry within the 3 km perifocal zone (1 household) and the 3–10 km surveillance zone (7 households) and detected no illness or death. We did not find any affected wildlife on site.
We collected oropharyngeal and cloacal swab samples from 8 birds to test for influenza A virus. We tested and subtyped the samples by real-time PCR at SENASA. We performed next-generation sequencing on positive samples as previously described (6) (Appendix 1). We deposited full and partial genome sequences in the GISAID database (https://gisaid.org; accession nos. EPI_ISL_19752381 and EPI_ISL_19823059–68).
Figure 1

Figure 1. Maximum-likelihood trees inferred for the 8 genome segments sequenced in this study of novel triple reassortant highly pathogenic avian influenza A(H5N1) virus, Argentina, 2025. Phylogenetic trees were inferred by using...
Figure 2
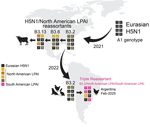
Figure 2. Key reassortment and migration events leading to the novel triple reassortant influenza A(H5N1) viruses in February 2025, Argentina. Each box represents 1 of the 8 segments of the influenza A...
Figure 3

Figure 3. Phylogenetic tree showing how North American low pathogenicity avian influenza (LPAI) lineage contributed nucleoprotein (NP) genes by reassortment to novel influenza A(H5N1) viruses from Argentina, 2025. We inferred the phylogenetic...
We inferred global avian influenza virus (AIV) phylogenies (including both HPAI and LPAI virus sequences) independently for each of the 8 gene segments to determine the genetic composition of H5N1 viruses from this outbreak (H5N1-Arg_Feb2025 viruses). The phylogenies indicated the H5N1-Arg_Feb2025 viruses are novel 4:3:1 triple reassortants (Figure 1; Appendix 1). Four gene segments (PB2, PB1, polymerase [PA], and nonstructural [NS]) belong to the South American LPAI lineage (Figure 1) that has circulated regionally in wild birds for decades (9–12). Three gene segments (hemagglutinin [HA], neuraminidase [NA], and matrix protein [MP]) clustered with H5N1 viruses of the B3.2 genotype and belong to the original Eurasian H5N1 lineage introduced to North America (Figure 2). We identified novel amino acid changes in the HA and NA segments of H5N1-Arg_Feb2025 viruses (Table) (13); the functional relevance of those changes is unknown but might merit further investigation if consistently detected in future outbreaks. One segment, nucleoprotein (NP), did not cluster with any previously known South American viruses and instead grouped with North American LPAI viruses (Figure 3). Because of limited surveillance in South America, where ≈90% of full-genome sequences are from Argentina and Chile, it was difficult to determine how the North America–derived NP segment became part of the triple reassortant H5N1 viruses we identified.
We have documented a reassortment event between HPAI H5N1 and endemic South America LPAI viruses. South American PB2 and PA segments are divergent from global AIV diversity (9) (Figure 1), indicating reassortment has expanded H5N1 polymerase diversity. Although the H5N1-Arg_Feb2025 viruses have exchanged 5 gene segments, they retained the original Eurasian MP segment (Figure 2), which remains conserved in most reassortant H5N1 viruses in North America. That segment conservation suggests the Eurasian MP segment might confer a selective advantage in HPAI H5 viruses. To date, we found no evidence of those novel 4:3:1 triple reassortant viruses in other South America countries; however, if future detections confirm wider spread, designation of a new H5N1 genotype would be warranted. Of consequence, genotyping tools such as the US Department of Agriculture’s GenoFlu should be expanded to include South American lineage genes for systematic classification of new virus genotypes.
Reassortment is a key mechanism in the evolution and host adaptation of AIV, often enabling their emergence in new animal communities and contributing to the development of strains with panzootic or pandemic potential (14,15). The genomic constellation of the H5N1-Arg_Feb2025 viruses parallels patterns found in North America, where clade 2.3.4.4b H5N1 viruses have also incorporated primarily internal LPAI genes. H5N1-Arg_Feb2025 viruses replaced the PB2, PB1, NP, and NS segments from the parental B3.2 genotype, a reassortment pattern observed in North American genotypes B3.6 and B3.13 (Figure 2), both derived from B3.2. However, the H5N1-Arg_Feb2025 viruses also replaced the Eurasian PA segment with a segment from South American LPAI viruses. Despite substantial genomic changes, the novel reassortant viruses from Argentina caused illness and death rates comparable to those previously observed for the B3.2 genotype. The primary clinical manifestation in this outbreak was diarrhea, affecting nearly two thirds of chickens. During H5N1 poultry outbreaks in Argentina in 2023, diarrhea was less common (reported in 29.7% of outbreaks), whereas high death rates (81.2%), lethargy (65.4%), cyanotic comb (57.4%), and neurologic signs (30.7%) were more frequent (n = 101 outbreaks) (SENASA, unpub. data). The predominance of gastrointestinal signs suggests possible shifts in tissue tropism or virulence. Also, the detection of a North American NP segment not previously identified in LPAI viruses from Argentina or elsewhere in South America highlights the need to strengthen regional AIV surveillance, even in the absence of active HPAI circulation. There is a lack of information on avian species and flyways involved in introductions of North American gene segments to South American LPAI viruses. Moreover, investigating the functional role of the North America–derived NP gene within the South American internal genes could clarify its potential contribution to the distinct gastrointestinal phenotype observed in this outbreak.
Further research on the diversity of LPAI viruses circulating in Neotropical wildlife will be essential to understand potential interactions between H5N1 and South American lineage strains and to assess the long-term consequences of the introduction of HPAI viruses into the region. Our findings underscore the critical importance of sustained influenza surveillance coupled with whole-genome sequencing to track the evolution of HPAI H5N1 and support efforts to control and mitigate its effect on domestic animals, wildlife, and human health.
Dr. Vanstreels is a veterinarian and an associate researcher with the Karen C. Drayer Wildlife Health Center at the University of California, Davis. His research interests include South American and Antarctic wildlife health, with a special interest in the ecology and effects of highly pathogenic avian influenza H5N1 on wild bird and marine mammal populations.
Acknowledgments
We thank the specific agreement between the Administración Nacional de Laboratorios e Institutos de Salud and the Servicio Nacional de Sanidad y Calidad Agroalimentaria in Argentina, which enabled the genome sequencing of the influenza virus strains detected.
This work was partially supported by the Department of Health and Human Services, Centers of Excellence for Influenza Research and Response (contract no. 75N93021C00014), and by the Intramural Research Program of the National Institutes of Health. Additional support for M.M.U. was provided by the US National Science Foundation Center for Pandemic Insights (award no. 2412522).
The contributions of the National Institutes of Health authors are considered Works of the United States Government.
References
- Uhart MM, Vanstreels RET, Nelson MI, Olivera V, Campagna J, Zavattieri V, et al. Epidemiological data of an influenza A/H5N1 outbreak in elephant seals in Argentina indicates mammal-to-mammal transmission. Nat Commun. 2024;15:9516.PubMedGoogle Scholar
- Pardo-Roa C, Nelson MI, Ariyama N, Aguayo C, Almonacid LI, Gonzalez-Reiche AS, et al. Cross-species and mammal-to-mammal transmission of clade 2.3.4.4b highly pathogenic avian influenza A/H5N1 with PB2 adaptations. Nat Commun. 2025;16:2232.PubMedGoogle Scholar
- Tawidian P, Torchetti MK, Killian ML, Lantz K, Dilione KE, Ringenberg JM, et al. Genotypic clustering of H5N1 avian influenza viruses in North America evaluated by ordination analysis. Viruses. 2024;16:1818.PubMedGoogle Scholar
- Nguyen TQ, Hutter CR, Markin A, Thomas M, Lantz K, Killian ML, et al. Emergence and interstate spread of highly pathogenic avian influenza A(H5N1) in dairy cattle in the United States. Science. 2025;388:
eadq0900 .PubMedGoogle Scholar - Ariyama N, Pardo-Roa C, Muñoz G, Aguayo C, Ávila C, Mathieu C, et al. Highly pathogenic avian influenza A(H5N1) clade 2.3.4.4b virus in wild birds, Chile. Emerg Infect Dis. 2023;29:1842–5.PubMedGoogle Scholar
- Artuso MC, Marchione VD, Benedetti E, Bonastre P, Alvarez AM, Piccini L, et al. Detection and characterization of highly pathogenic avian influenza A (H5N1) clade 2.3.4.4b virus circulating in Argentina in 2023. Rev Argent Microbiol. 2024:S0325-7541(24)00115-9.
- Marandino A, Tomás G, Panzera Y, Leizagoyen C, Pérez R, Bassetti L, et al. Spreading of the high-pathogenicity avian influenza (H5N1) virus of clade 2.3.4.4b into Uruguay. Viruses. 2023;15:1906.PubMedGoogle Scholar
- Sevilla N, Lizarraga W, Jimenez-Vasquez V, Hurtado V, Molina IS, Huarca L, et al. Highly pathogenic avian influenza A (H5N1) virus outbreak in Peru in 2022-2023. Infect Med (Beijing). 2024;3:
100108 .PubMedGoogle Scholar - Rimondi A, Gonzalez-Reiche AS, Olivera VS, Decarre J, Castresana GJ, Romano M, et al. Evidence of a fixed internal gene constellation in influenza A viruses isolated from wild birds in Argentina (2006-2016). Emerg Microbes Infect. 2018;7:194.PubMedGoogle Scholar
- Jiménez-Bluhm P, Karlsson EA, Freiden P, Sharp B, Di Pillo F, Osorio JE, et al. Wild birds in Chile Harbor diverse avian influenza A viruses. Emerg Microbes Infect. 2018;7:44.PubMedGoogle Scholar
- Nelson MI, Pollett S, Ghersi B, Silva M, Simons MP, Icochea E, et al. The genetic diversity of influenza A viruses in wild birds in Peru. PLoS One. 2016;11:
e0146059 .PubMedGoogle Scholar - Araujo AC, Cho AY, Silva LMN, Port D, Demoliner M, Gularte JS, et al. Whole genome sequencing of low pathogenicity avian influenza virus (H6N2) detected from a Brazilian teal (Amazonnetta brasiliensis) in Brazil, 2023. Microbiol Resour Announc. 2024;13:
e0015824 .PubMedGoogle Scholar - Carter T, Iqbal M. The influenza A virus replication cycle: a comprehensive review. Viruses. 2024;16:316.PubMedGoogle Scholar
- Dugan VG, Chen R, Spiro DJ, Sengamalay N, Zaborsky J, Ghedin E, et al. The evolutionary genetics and emergence of avian influenza viruses in wild birds. PLoS Pathog. 2008;4:
e1000076 .PubMedGoogle Scholar - Youk S, Torchetti MK, Lantz K, Lenoch JB, Killian ML, Leyson C, et al. H5N1 highly pathogenic avian influenza clade 2.3.4.4b in wild and domestic birds: Introductions into the United States and reassortments, December 2021-April 2022. Virology. 2023;587:
109860 .PubMedGoogle Scholar
Figures
Table
Cite This ArticleOriginal Publication Date: December 12, 2025
Table of Contents – Volume 31, Number 12—December 2025
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Agustina Rimondi, Robert Koch-Institut, Seestr. 10, 13353, Berlin, Germany
Top