Volume 31, Number 12—December 2025
Dispatch
Novel Highly Pathogenic Avian Influenza A(H5N1) Virus, Argentina, 2025
Figure 1
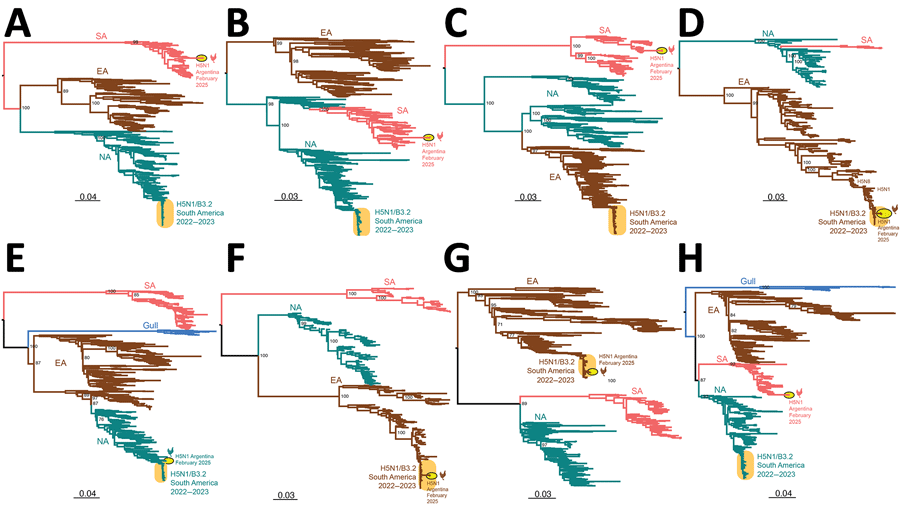
Figure 1. Maximum-likelihood trees inferred for the 8 genome segments sequenced in this study of novel triple reassortant highly pathogenic avian influenza A(H5N1) virus, Argentina, 2025. Phylogenetic trees were inferred by using IAVs collected globally in avian hosts. A) Gene segment PB2. B) Gene segment PB1. C) Gene segment PA. D) Gene segment H5. E) Gene segment NP. F) Gene segment N1. G) Gene segment MP. H) Gene segment NS; only A allele is shown. The number of sequences used to construct each tree was 676 for PB2, 667 for PB1, 686 for PA, 271 for H5, 682 for NP, 443 for N1, 639 for MP, and 506 for NS (Appendix 2, https://wwwnc.cdc.gov/EID/article/31/12/25-0783-App2.xlsx). Trees are midpoint rooted for clarity. Key node bootstrap values are shown. Teal shading represents NA lineage, brown shading represents EA lineage, pink shading represents SA lineage, and blue shading represents gull lineage. Orange oval represents previously reported SA H5N1 clade B3.2 genotype virus. Yellow oval with black outline and chicken silhouette represents the novel H5N1 virus found in Argentina. Branch lengths are drawn to scale. Scale bars represent nucleotide substitutions per site. EA, Eurasian; MP, matrix protein; NA, North American; NP, nucleoprotein; NS, nonstructural protein; PA, polymerase; PB, polymerase basic; SA, South American.