Volume 31, Number 7—July 2025
Dispatch
Borrelia Lineages Adjacent to Zoonotic Clades in Black Flying Foxes (Pteropus alecto), Australia, 2018–2020
Cite This Article
Citation for Media
Abstract
We explored the role of black flying foxes (Pteropus alecto) in Australia as reservoirs of Borrelia bacteria. We found bats infected with 2 Borrelia haplotypes phylogenetically distinct from Lyme or relapsing fever clades. Efforts to sample black flying foxes and their ectoparasites are needed to evaluate zoonotic potential of those Borrelia lineages.
Bacteria in the genus Borrelia are causative agents of 2 diseases of substantial public health concern, Lyme borreliosis and relapsing fever. Lyme borreliosis is the most frequently reported vectorborne infection in the Northern Hemisphere; its vector is Ixodidae family ticks (1). By contrast, relapsing fever is globally distributed; its vector is predominantly Argasidae family ticks, and thousands of cases of febrile illness in humans are attributed to it annually (2).
Two of the 3 well-recognized monophyletic Borrelia clades are the B. burgdorferi sensu lato complex that causes Lyme borreliosis and the relapsing fever group that causes relapsing fever; the third clade is a group hosted by reptiles and echidnas (3). Borrelia from the B. burgdorferi s.l. and relapsing fever clades are harbored by a broad range of vertebrate hosts, including birds, reptiles, and mammals. Identifying reservoir hosts and clarifying their role in propagating Borrelia are critical for monitoring and mitigating spillover risk. For example, migratory birds can contribute to the long-distance dispersal of B. burgdorferi s.l. by transporting millions of ticks within and across continents (4).
Bats might play an underrecognized role in the dispersal and enzootic maintenance of Borrelia bacteria. Bats are volant mammals that have been known for more than a century to harbor borrelial spirochaetes (5), and surveys within the last 5 years indicate bats can host Borrelia spp. from the relapsing fever group and from a clade adjacent to B. burgdorferi s.l. (6,7). Evidence has shown that bat-associated Borrelia infections can be zoonotic because Borrelia lineages recovered from bats and bat ticks have been implicated in febrile illness in humans (8). Therefore, the expansion of Borrelia research in chiropteran hosts could provide more information about the current and future welfare of both bat and human populations.
Pteropodidae family flying foxes (Pteropus spp. bats) represent a group that is highly prominent at the human–wildlife interface in Australia and therefore a target for Borrelia bacteria surveillance. The increasing propensity for flying foxes to roost in or near human settlements could increase the overlap of humans and bat ectoparasites and create a potential Borrelia bacteria spillover pathway (9,10). Although black flying foxes (P. alecto) have been the subject of extensive research as reservoir hosts of Hendra virus, relatively little study has been devoted to their bacterial communities, including those that could be pathogenic (11). We assessed the presence, diversity, and phylogenetic placement of Borrelia spp. associated with black flying foxes in Australia.
Figure 1
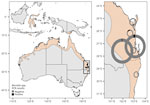
Figure 1. Sampling sites in eastern Australia (Queensland and New South Wales) of Borrelia lineages adjacent to zoonotic clades in black flying foxes (Pteropus alecto), Australia, 2018–2020. ...
As part of ongoing research on the ecology of Hendra virus, we collected blood samples from 840 black flying foxes across 6 sites in southern Queensland and northern New South Wales, Australia, during May 2018–September 2020 (Figure 1). We captured bats with mist nets, anesthetized them with isoflurane, and took blood samples and preserved on Whatman FTA cards (QIAGEN, https://www.qiagen.com) until further processing (12). The Montana State University Institutional Animal Care and Use Committee (approval no. 201750, https://www.montana.edu/ric/iacuc/iacuc-committee.html) and Griffith University Animal Ethics Committee (approval nos. ENV/10/16/AEC and ENV/07/20/AEC, https://www.griffith.edu.au/research/research-services/research-ethics-integrity/animal/animal-ethics-committee) approved the field protocols.
We used QIAamp DNA Investigator Kits (QIAGEN) to extract genomic DNA from blood (four to five 2-mm punches per sample), according to the manufacturer’s instructions. To determine Borrelia spp. infection, we used PCR targeting of the 16S rRNA gene, flagellin (flaB) gene, and 16S–23S rRNA intergenic spacer (IGS) (Appendix Table 1). We purified PCR products within the expected size range for positive results with the Wizard SV Gel and PCR Clean-Up System (Promega, https://www.promega.com) and had the products Sanger sequenced in both directions at Eurofins Genomics (https://ww.eurofinsgenomics.com). We report prevalence data for IGS, but that gene was not included in phylogenetic analyses because of insufficient reference sequences in GenBank. We manually edited and trimmed the Borrelia spp. sequences from black flying foxes and aligned those sequences with reference sequences in GenBank by using the MUSCLE algorithm in Geneious version 2024.0.5 (https://www.geneious.com).
We constructed single-marker phylogenies by using both Bayesian and maximum-likelihood methods because too few bat-associated reference sequences of either flaB or 16S were available in GenBank to provide an informative concatenated analysis. For Bayesian analyses, we used MrBayes version 3.2 (https://nbisweden.github.io/MrBayes/download.html) with 10 million Markov chain Monte Carlo generations and a default burn-in of 25%. For maximum-likelihood analyses, we used RAxML version 8.0 (https://github.com/amkozlov/raxml-ng) and a starting tree obtained by searching for the best-scoring maximum-likelihood tree in a single run and calculating branch support with 1,000 rapid bootstrap replicates. For all trees, we used a general time reversible with a proportion of invariable sites and gamma distribution nucleotide substitution model (13).
Figure 2
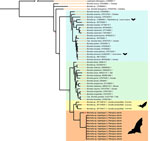
Figure 2. Bayesian phylogenetic tree of flaB gene in a study of Borrelia lineages adjacent to zoonotic clades in black flying foxes (Pteropus alecto), Australia, 2018–2020....
Figure 3
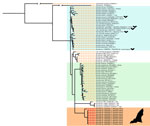
Figure 3. Bayesian phylogenetic tree 16S rRNA gene in a study of Borrelia lineages adjacent to zoonotic clades in black flying foxes (Pteropus alecto), Australia, 2018–2020. The tree...
Across our ≈2-year study period and 6 roosts, 17 (2% [95% CI 1.3%–3.2%]) of 840 black flying foxes tested positive for Borrelia spp. by using PCR (Table). Roost-specific infection prevalence ranged from 0 of 20 samples (no infection found in 3 of the 6 roosts) to 3 (15% [95% CI 5.2%–36.0%]) of 20 samples in Gympie, Queensland, Australia. The roosts with greatest sampling volume were in Toowoomba (402 bats sampled, 3 [0.7%] infected) and Redcliffe (374 bats sampled, 11 [2.9%] infected) in Queensland, Australia (Appendix Table 2). Most (13/17) infected bats yielded usable sequence data from either 16S rRNA or flaB gene targets, which represented 2 haplotypes (p-distances = 8.2% for flaB and 1.3% for 16S rRNA). Topologies were similar across tree-building methods (Appendix Figures 1, 2) but were somewhat discordant between gene targets. The flaB phylogeny grouped black flying fox Borrelia spp. with lineages from Macaregua Cave in Colombia in a clade sister to B. burgdorferi s.l. (Figure 2), but 16S rRNA sequence data are not available from Macaregua Cave as of 2025, and no other sequences from bats in Australia are available. The 16S rRNA phylogeny (Figure 3) was also less effective at resolving relationships across the relapsing fever and B. burgdorferi s.l. groups supported in previous analyses using multiple markers (3). All sequences included here are available at GenBank (accession nos. PQ488732–41 [16S rRNA], PQ492350–60 [flaB], and PQ490736–46 [16S–23S IGS]).
This study corroborated that bats can host Borrelia infections evolutionarily distinct from recognized clades. However, the clades are more closely related to B. burgdorferi s.l. than the relapsing fever group. We recovered sequences from 11 of 17 infected bats in 2 Borrelia spp. lineages (Figure 2). Haplotype 1 was represented by a single host that shared a roost in Gympie with a bat infected with haplotype 2, suggesting that variation is unlikely to be structured by geographic isolation. Phylogenetic reconstruction of flaB and 16S rRNA suggested Borrelia infections from black flying foxes belong to a clade adjacent to existing B. burgdorferi s.l. and that relapsing fever groups and are most closely related to Borrelia spp. hosted by phyllostomid bats (Chiroptera order) in Colombia (7).
Our results are a base for establishing the presence and phylogenetic placement of Borrelia infections in flying foxes but underscore research gaps in characterizing their zoonotic potential. Virulence of those lineages in flying foxes is unknown. Although bats are tolerant of multiple viruses, lethal Borrelia infections in bats are documented (14). The arthropod vector of lineages described in this study also remains unidentified, but host specificity and geographic range of that vector should strongly influence zoonotic risk. For example, certain ixodid bat ticks have generalist feeding habits that could position them as an epidemiologic link between bats, domestic animals, and humans (15). Targeted efforts to sample black flying foxes and their ectoparasites across their range are needed to clarify information regarding the zoonotic potential of the novel Borrelia lineages described in this study.
Ms. Verrett is a doctoral candidate at the University of Oklahoma School of Biological Sciences. Her primary research interests are in how host–parasite interactions are influenced by seasonal movement and anthropogenic change, particularly in migratory bird systems.
Acknowledgments
We thank the Butchulla, Gumbainggir, Kabi Kabi, Turrbal, and Yuggera Ugarapul people, who are the Traditional Custodians of the land upon which this work was conducted. We also thank government and private landholders for granting permission for fieldwork and broader team members and volunteers for their contributions, including Peggy Eby, Maureen Kessler, Liam Chirio, Mandy Allonby, Rachael Smethurst, Remy Brooks, Liam McGuire, Kirk Silas, Ticha Padgett-Stewart, Denise Karkkainen, Justine Scaccia, Ariana Ananda, Emma Glennon, Hannah Eiseman, Sara LaTrielle, Isaac Knights, Dian Riseley, and Stella Maris Januario da Silva.
Research was conducted with a Scientific Purposes Permits from the Queensland Department of Environment and Heritage Protection (permit nos. WISP17455716 and WA0012532), a permit to Take, Use, Keep or Interfere with Cultural or Natural Resources (Scientific Purpose) from the Department of National Parks, Sport and Racing (permit no. WITK18590417), a Scientific License from the New South Wales Parks and Wildlife Service (permit no. SL101800), and a general and products liability protection permit (permit no. GRI 18 GPL) and with permission to undertake research on council and private land. Sample import to Montana State University was authorized by import permit no. 20200728-2504A.
This work was funded by the Defense Advanced Research Projects Agency (DARPA) Preventing Emerging Pathogenic Threats (PREEMPT) program (cooperative agreement no. D18AC00031) and the US National Science Foundation (grant nos. DEB-1716698, EF-2133763, and EF-2231624). The content of the information does not necessarily reflect the position or the policy of the US government, and no official endorsement should be inferred. Molecular analyses were also partially supported by the Early Career Grants Programme of the Royal Society of Tropical Medicine and Hygiene.
This article was preprinted at https://www.biorxiv.org/content/10.1101/2024.12.20.629695v1.
References
- Khatchikian CE, Prusinski MA, Stone M, Backenson PB, Wang I-N, Foley E, et al. Recent and rapid population growth and range expansion of the Lyme disease tick vector, Ixodes scapularis, in North America. Evolution. 2015;69:1678–89. DOIPubMedGoogle Scholar
- Jakab Á, Kahlig P, Kuenzli E, Neumayr A. Tick borne relapsing fever–a systematic review and analysis of the literature. PLoS Negl Trop Dis. 2022;16:
e0010212 . DOIPubMedGoogle Scholar - Loh S-M, Gillett A, Ryan U, Irwin P, Oskam C. Molecular characterization of ‘Candidatus Borrelia tachyglossi’ (family Spirochaetaceae) in echidna ticks, Bothriocroton concolor. Int J Syst Evol Microbiol. 2017;67:1075–80. DOIPubMedGoogle Scholar
- Ogden NH, Lindsay LR, Hanincová K, Barker IK, Bigras-Poulin M, Charron DF, et al. Role of migratory birds in introduction and range expansion of Ixodes scapularis ticks and of Borrelia burgdorferi and Anaplasma phagocytophilum in Canada. Appl Environ Microbiol. 2008;74:1780–90. DOIPubMedGoogle Scholar
- Nicolle C, Comte C. On a spirillosis of a cheiroptera (Vespertilio kuhli) [in French]. Ann. de l’Inst. Pasteur 1906;20:311–20.
- Colunga-Salas P, Sánchez-Montes S, León-Paniagua L, Becker I. Borrelia in neotropical bats: detection of two new phylogenetic lineages. Ticks Tick Borne Dis. 2021;12:
101642 . DOIPubMedGoogle Scholar - Muñoz-Leal S, Faccini-Martínez ÁA, Pérez-Torres J, Chala-Quintero SM, Herrera-Sepúlveda MT, Cuervo C, et al. Novel Borrelia genotypes in bats from the Macaregua Cave, Colombia. Zoonoses Public Health. 2021;68:12–8. DOIPubMedGoogle Scholar
- Qiu Y, Nakao R, Hang’ombe BM, Sato K, Kajihara M, Kanchela S, et al. Human borreliosis caused by a New World relapsing fever Borrelia-like organism in the old world. Clin Infect Dis. 2019;69:107–12. DOIPubMedGoogle Scholar
- Eby P, Peel AJ, Hoegh A, Madden W, Giles JR, Hudson PJ, et al. Pathogen spillover driven by rapid changes in bat ecology. Nature. 2023;613:340–4. DOIPubMedGoogle Scholar
- Szentiványi T, Takács N, Sándor AD, Péter Á, Boldogh SA, Kováts D, et al. Bat-associated ticks as a potential link for vector-borne pathogen transmission between bats and other animals. PLoS Negl Trop Dis. 2024;18:
e0012584 . DOIPubMedGoogle Scholar - Szentivanyi T, McKee C, Jones G, Foster JT. Trends in bacterial pathogens of bats: global distribution and knowledge gaps. Transbound Emerg Dis. 2023;2023:
9285855 . DOIPubMedGoogle Scholar - Hansen D, Hunt BE, Falvo CA, Ruiz-Aravena M, Kessler MK, Hall J, et al.; Bat One Health. Morphological and quantitative analysis of leukocytes in free-living Australian black flying foxes (Pteropus alecto). PLoS One. 2022;17:
e0268549 . DOIPubMedGoogle Scholar - Abadi S, Azouri D, Pupko T, Mayrose I. Model selection may not be a mandatory step for phylogeny reconstruction. Nat Commun. 2019;10:934. DOIPubMedGoogle Scholar
- Irving AT, Ahn M, Goh G, Anderson DE, Wang L-F. Lessons from the host defences of bats, a unique viral reservoir. Nature. 2021;589:363–70. DOIPubMedGoogle Scholar
- Evans NJ, Bown K, Timofte D, Simpson VR, Birtles RJ. Fatal borreliosis in bat caused by relapsing fever spirochete, United Kingdom. Emerg Infect Dis. 2009;15:1331–3. DOIPubMedGoogle Scholar
Figures
Table
Cite This ArticleOriginal Publication Date: May 21, 2025
Table of Contents – Volume 31, Number 7—July 2025
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Taylor B. Verrett, University of Oklahoma School of Biological Sciences, 730 Van Vleet Oval, Norman, OK 73019, USA
Top