Volume 31, Number 9—September 2025
Dispatch
Novel Henipavirus, Salt Gully Virus, Isolated from Pteropid Bats, Australia
Figure 1
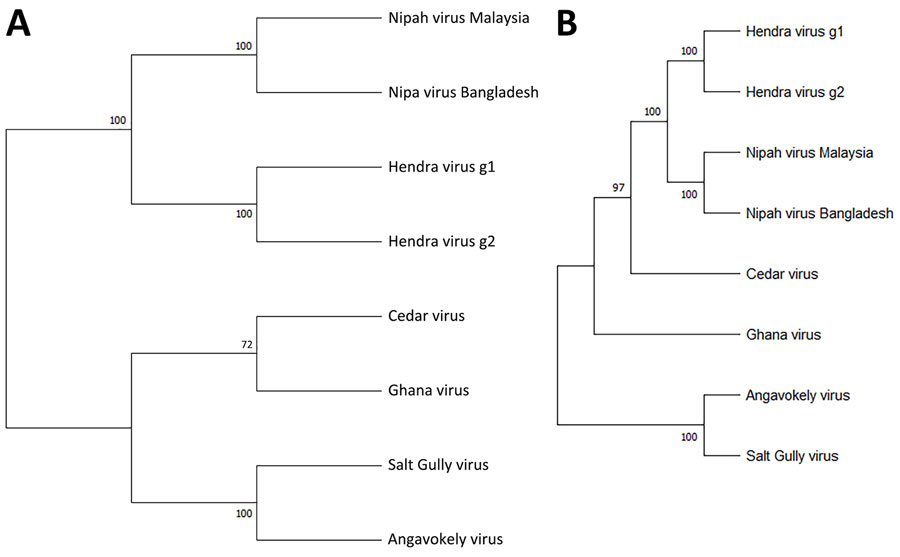
Figure 1. Phylogenetic analysis of members of the genus Henipavirus from a study investigating a novel henipavirus, Salt Gully virus, isolated from pteropid bats, Australia. A) We aligned complete L protein amino acid sequences by using ClustalW (https://www.genome.jp/tools-bin/clustalw) and inferred evolutionary history by using the maximum-likelihood method and the Jones-Taylor-Thornton matrix-based model. B) We aligned complete virus genome sequences by Muscle software and inferred evolutionary history by using the maximum-likelihood method and general time reversible plus gamma plus invariate sites model. Bootstrap support values (1,000 replicates) are shown next to each branching node. Evolutionary analyses were conducted in MEGA11 (https://www.megasoftware.net).