Volume 32, Number 4—April 2026
Dispatch
Cardiomyopathy Caused by Coxsackievirus Strain A9 in Previously Healthy Child, Northeastern France, 2024
Figure 2
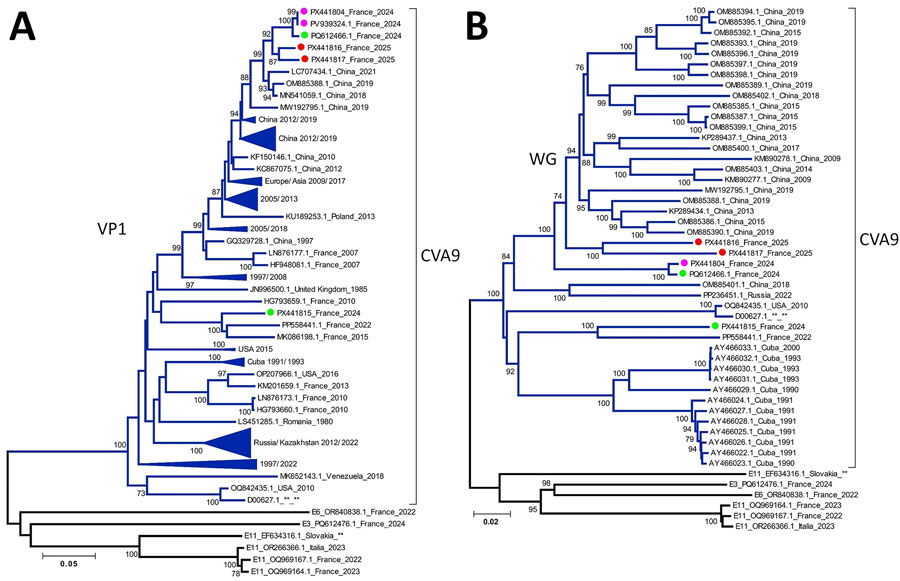
Figure 2. Phylogenetic analysis of CVA9 sequences from samples collected from a pediatric patient with severe inflammatory cardiomyopathy caused by CVA9, northeastern France, 2024, and reference sequences. A) All available CVA9 sequences from GenBank containing the complete VP1 (1D) gene (with a maximum of 25 missing bases at either end) aligned with CVA9 sequences obtained in this study and in France during 2024–2025. B) All available CVA9 sequences from GenBank containing the complete genome sequence (entire open reading frame with <60 bases missing from the 5′ UTR) aligned with CVA9 sequences obtained in this study and in France during 2024–2025, along with selected echovirus reference strains. We conducted sequence alignment by using BioEdit version 7.2.5 (https://thalljiscience.github.io/page2.html) and constructed phylogenetic trees by using the neighbor-joining method and the Tamura-Nei substitution model in MEGA 5 (http://www.megasoftware.net) We assessed the robustness of tree nodes with 1,000 bootstrap replicates; only bootstrap values >70% are shown. Pink circles indicate sequences generated in this study, red circles indicate CVA9 isolates from France in 2025, and green circles indicate CVA9 isolates from France in 2024. Sequences with close phylogenetic relationships were grouped into clusters, annotated with the country of origin and the range of isolation years. Clusters without specific geographic annotation contained sequences isolated from multiple continents. Branch numbers indicate bootstrap values. Scale bars represent substitutions per site. CVA9, coxsackievirus A9; VP, viral protein; WG, whole-genome.
1These authors contributed equally to this article.