Volume 32, Number 7—July 2026
Research Letter
Ancylostoma ceylanicum Hookworm, Rural Papua New Guinea, 2020
Figure
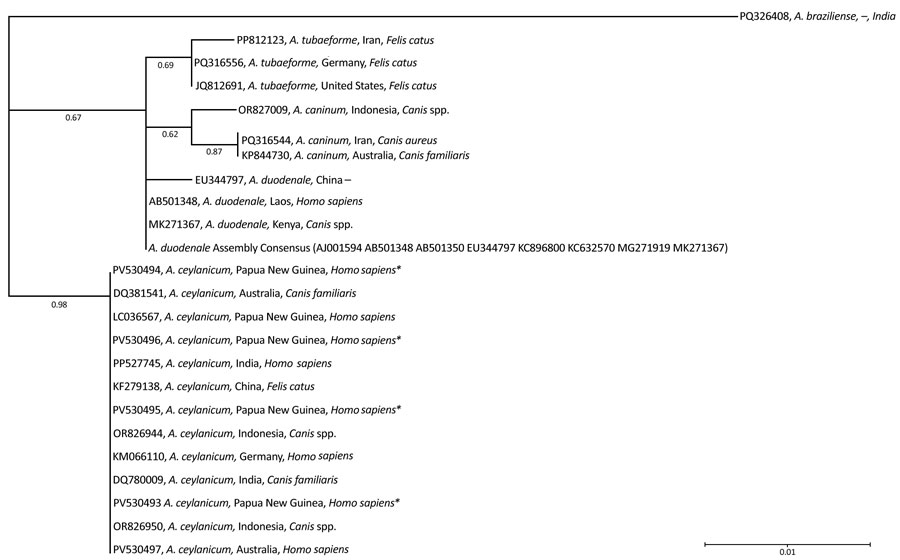
Figure. Phylogenetic analysis of Ancylostoma spp. in study of A. ceylanicum hookworm, rural Papua New Guinea, 2020. Analysis is based on the internal transcribed spacer sequence regions 1 and 2 and conducted with the maximum-likelihood method, using the Kimura 2-parameter model. Model selection was based on penalized-likelihood information criteria. The phylogenetic tree was visually adjusted using Interactive Tree of Life version 7.2.2 (https://itol.embl.de). Values at branch nodes indicate bootstrap support values (1,000 replicates). A. braziliense was used as an outgroup to root the tree. GenBank accession numbers are shown; asterisks (*) denote sequences identified in this study (accession nos. PV530493–96). We used A. ceylanicum (accession no. PV530497) as a positive control. Dash (–) indicates that no information was available for a particular attribute of the strain. Scale bar represents 0.01 substitutions per site.