Volume 4, Number 1—March 1998
Synopsis
Proteases of Malaria Parasites: New Targets for Chemotherapy
Cite This Article
Citation for Media
Abstract
The increasing resistance of malaria parasites to antimalarial drugs is a major contributor to the reemergence of the disease as a major public health problem and its spread in new locations and populations. Among potential targets for new modes of chemotherapy are malarial proteases, which appear to mediate processes within the erythrocytic malarial life cycle, including the rupture and invasion of infected erythrocytes and the degradation of hemoglobin by trophozoites. Cysteine and aspartic protease inhibitors are now under study as potential antimalarials. Lead compounds have blocked in vitro parasite development at nanomolar concentrations and cured malaria-infected mice. This review discusses available antimalarial agents and summarizes experimental results that support development of protease inhibitors as antimalarial drugs.
Hundreds of millions of cases of malaria occur annually, and infections with Plasmodium falciparum, the most virulent human malaria parasite, cause more than one million deaths per year (1). Despite extensive control efforts, the incidence of the disease is not decreasing in most malaria-endemic areas of the world, and in some it is clearly increasing (2). Malaria also remains a major risk to travelers from industrialized to developing countries. Because malaria parasites are increasingly resistant to antimalarial drugs, appropriately counseled travelers to malaria-endemic regions are more likely to contract malaria now than they were 40 years ago.
Malaria control efforts include attempts to develop an effective vaccine, eradicate mosquito vectors, and develop new drugs (2,3). However, the development of a vaccine has proven very difficult, and a highly effective vaccine will probably not be available in the near future (4). Efforts to control Anopheles mosquitoes have had limited success, although the use of insecticide-impregnated bed nets does appear to reduce malaria-related death rates (5). In addition, methods to replace natural vector populations with mosquitoes unable to support parasite development are under study and may contribute to malaria control in the long term (6). However, the current limitations of vaccine and vector control, as well as the increasing resistance of malaria parasites to existing drugs, highlight the continued need for new antimalarial agents.
Antimalarial drugs have been used for centuries. Early natural products, including the bark of the cinchona tree in South America and extracts of the wormwood plant in China, were among the first effective antimicrobial agents to be used. Cinchona bark was used in Europe beginning in the 17th century, and upon its isolation from bark in 1820, quinine became widely used. In the last 50 years, extensive efforts, including the screening of hundreds of thousands of compounds, have led to the development of a number of effective synthetic antimalarial drugs. The most important of these, chloroquine, has been the mainstay of antimalarial chemotherapy for the last 50 years. The compound eradicates parasites rapidly, has minimal toxicity, is widely available at low cost throughout the world, and needs to be taken only once a week for chemoprophylaxis. However, resistance to chloroquine has been steadily increasing since the drug's initial use in South America and Southeast Asia in the late 1950s. Chloroquine resistance is now widespread in most P. falciparum-endemic areas of the world (3). Thus, the use of chloroquine for presumptive treatment of falciparum malaria or for chemoprophylaxis is usually no longer appropriate (7). Moreover, resistance to chloroquine of P. vivax, the second most lethal human malaria parasite, is increasing in South Asia (8).
No other antimalarial drug (9-12) is as efficacious and safe as chloroquine (Table 1). The best antimalarial drug for treating chloroquine-resistant falciparum malaria remains quinine (or intravenous quinidine), which is fairly toxic; quinine resistance is increasing in Southeast Asia, particularly in the border areas of Thailand (9). Amodiaquine, used to treat chloroquine-resistant malaria in developing countries, is also quite toxic, and resistance to it is also common (13">13). Mefloquine (14) is widely used for chemoprophylaxis against chloroquine-resistant P. falciparum, but its use is limited by toxicity (15) and (in the developing world) high cost. Mefloquine is not approved for treatment of malaria in the United States because of the neurotoxicity of doses required for the treatment. Fansidar, a combination of sulfadoxine and pyrimethamine, is no longer recommended for chemoprophylaxis because of its dermatologic toxicity (15). Fansidar is also not an ideal drug for treatment because it is slow acting, but it is increasingly important in treating chloroquine-resistant malaria in developing countries because economic constraints limit the use of other agents (16). The use of both mefloquine and Fansidar will increasingly be limited by drug resistance, already widespread in parts of Southeast Asia (9,17).
Other antimalarial drugs have specialized uses. Tetracyclines and some other antibiotics (clindamycin, sulfas) are slow acting and generally best used as an adjunct to quinine therapy in treating falciparum malaria (9). Doxycycline is also used for chemoprophylaxis in regions with high levels of drug resistance, especially Southeast Asia (10,17). Other drugs for chemoprophylaxis include proguanil, which remains effective in combination with chloroquine in many areas other than Southeast Asia, and Maloprim, a combination of dapsone and pyrimethamine (10,17). Resistance to these drugs is fairly common, however. Primaquine has a well-defined specific role: eradicating chronic liver stages of P. vivax and P. ovale after treating the acute blood infection with chloroquine.
Relatively few new antimalarial drugs are undergoing clinical testing (Table 2). Halofantrine, identified in the 1940s, was not developed until the 1980s; its use has been limited by variable oral absorption and cardiac toxicity (18). The drug is approved in the United States for treatment of chloroquine-resistant <I>P. falciparum</I> infection, although in most cases quinine (or intravenous quinidine) is preferable. The most effective new drugs are artemisinin and related compounds. Artemisinin was isolated in 1972 from <I>Artemisia annua</I>, a plant used in China for centuries to treat fever (19). Artemisinin derivatives (artesunate, artelinate, artemether, arteether, dihydroartemisinin) have been synthesized and are undergoing extensive clinical testing. These compounds, which are already widely used in some areas, are potent, rapidly acting antimalarials that are effective against chloroquine-resistant P. falciparum (20). Because recrudescences of infection after treatment are common, however, artemisinin and related compounds might best be used in combination with another drug.
Other compounds are under evaluation. Atovaqone (21), which is approved for treating patients with Pneumocystis infections, appears to be effective against malaria in combination with proguanil (22), but its use has been limited by recrudescence after treatment. Pyronaridine, an acridine derivative used to treat malaria in China, has shown efficacy against falciparum malaria (23). The iron chelator desferrioxamine enhances the clearance of parasites in mild malaria (24) and, in conjunction with quinine and Fansidar, hastens recovery from deep coma in severe falciparum malaria (25). Azithromycin, a quinolone antibiotic, appears efficacious in malaria chemoprophylaxis (26).
Figure
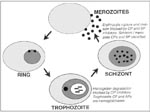
Figure. Protease targets in erythrocytic malaria parasites. The Plasmodium falciparum erythrocytic life cycle is shown schematically, and data supporting cysteine (CP), serine (SP), and aspartic (AP) proteases of the different parasite stages...
The limitations of antimalarial chemotherapy underscore the need for new drugs, ideally directed against new targets. Potential targets for chemotherapy include malarial proteases (27). The erythrocytic life cycle, which is responsible for all clinical manifestations of malaria, begins when free merozoites invade erythrocytes. The intraerythrocytic parasites develop from small ring-stage organisms to larger, more metabolically active trophozoites and then to multinucleated schizonts. The erythrocytic cycle is completed when mature schizonts rupture erythrocytes, releasing numerous invasive merozoites. Proteases appear to be required for the rupture and subsequent reinvasion of erythrocytes by merozoite-stage parasites and for the degradation of hemoglobin by intraerythrocytic trophozoites (Figure).
The rupture of erythrocytes by mature schizonts and the subsequent invasion of erythrocytes by free merozoites appear to require malarial protease activity, possibly to breach the erythrocyte cytoskeleton, a complex network of proteins. In addition, a number of malarial proteins are proteolytically processed during the late schizont and merozoite life-cycle stages; for example, merozoite surface protein-1 is processed in a manner inhibited by serine protease inhibitors (28), presumably to facilitate the complex series of events involved in erythrocyte rupture and invasion (29). Although the specific roles of different classes of proteases are not completely clear, inhibitors of cysteine and serine proteases have consistently blocked erythrocyte rupture and invasion (27).
Candidate P. falciparum rupture/invasion proteases have been identified, but none has been fully characterized biochemically or molecularly: 1) a 68 kD cysteine protease was identified in schizonts and merozoites and localized to the merozoite apex, suggesting that it may be released from the rhoptry organelle during invasion (30); 2) a cysteine protease of mature schizonts and a serine protease of merozoites were identified in highly synchronized parasites (31); 3) a serine protease was shown to be bound to the schizont/merozoite membrane by a glycosyl-phosphatidylinositol anchor, to be activated by phosphatidylinositol-specific phospholipase C during the merozoite stage, and to be capable of cleaving the erythrocyte cytoskeletal protein band 3 (32,33); 4) another protease, inhibited by both cysteine and serine protease inhibitors, hydrolyzed the erythrocyte cytoskeletal proteins spectrin and band 4.1 (34); and 5) the serine repeat antigen (35,36) and the related protein SERP H (37), both expressed in mature schizonts, have important similarities in their sequences with cysteine proteases. Further research should identify the specific biologic roles of the proteases mentioned and better characterize these enzymes, thus fostering the development of specific inhibitors.
Host proteases may also play a role in erythrocyte rupture by P. falciparum. In recent studies, host urokinase was shown to bind to the surface of P. falciparum-infected erythrocytes, and the depletion of urokinase from parasite culture medium inhibited erythrocyte rupture by mature schizonts (38). This inhibition was reversed by exogenous urokinase.
Drug Development Efforts
Synthetic peptide inhibitors of the P. falciparum schizont cysteine protease Pf 68 inhibited erythrocyte invasion by cultured parasites (39,40). The most effective peptide, GlcA-Val-Leu-Gly-Lys-NHC2H5, inhibited the protease and blocked parasite development at high micromolar concentrations (40; Table 3). Although these results do not demonstrate levels of inhibition expected to be therapeutically relevant, they suggest that a specific protease activity is required for erythrocyte invasion by malaria parasites and thus is a potential target for antimalarial drugs.
Extensive evidence suggests that the degradation of hemoglobin is necessary for the growth of erythrocytic malaria parasites, apparently to provide free amino acids for parasite protein synthesis (27,50). In P. falciparum, hemoglobin degradation occurs predominantly in trophozoites and early schizonts, the stages at which the parasites are most metabolically active. Trophozoites ingest erythrocyte cytoplasm and transport it to a large central food vacuole. In the food vacuole, hemoglobin is broken down into heme, a major component of malarial pigment (51), and globin, which is hydrolyzed to its constituent amino acids. The food vacuole is an acidic organelle analogous to lysosomes. Several lysosomal proteases are well characterized, including cysteine (cathepsins B, H, and L) and aspartic (cathepsin D) proteases (52), and malaria parasites contain analogous food vacuole proteases that degrade hemoglobin. At least two aspartic proteases and one cysteine protease have been isolated from purified P. falciparum food vacuoles (53).
Malarial aspartic protease activities have been identified (54-60). Two recently characterized aspartic proteases (plasmepsin I and plasmepsin II) are located in the food vacuole, have acid pH optima, and share sequence homology with other aspartic proteases (41,53,61,62). Furthermore, the aspartic proteases can cleave hemoglobin. One of the enzymes, plasmepsin I, cleaves native hemoglobin (53,59). Plasmepsin II appears to prefer denatured globin as a substrate (53). On the basis of these data, plasmepsin I is thought to be responsible for initial cleavages of hemoglobin after the molecule is transported to the food vacuole (53).
Incubation of cultured P. falciparum parasites with the protease inhibitor leupeptin caused trophozoite food vacuoles to fill with apparently undegraded erythrocyte cytoplasm (63-65). Analysis of the leupeptin-treated parasites showed that they contained large quantities of undegraded globin, while minimal globin was detectable in control parasites (64,66). Leupeptin inhibits both cysteine and some serine proteases, but the highly specific cysteine protease inhibitor E-64 also caused undegraded globin to accumulate. After parasites were incubated with inhibitors of other classes of proteases including the aspartic protease inhibitor pepstatin (63-67), globin did not accumulate. More recent studies that used nondenaturing electrophoretic methods demonstrated that cysteine protease inhibitors not only blocked malarial globin hydrolysis, but also inhibited earlier steps in hemoglobin degradation, including denaturation of the hemoglobin tetramer and the release of heme from globin (68). Another study showed that E-64, but not pepstatin, inhibited the production of hemozoin (the malarial end product of heme) by cultured parasites (69). These results suggest that a cysteine protease is required for initial steps in hemoglobin degradation by P. falciparum.
A P. falciparum trophozoite cysteine protease with biochemical features expected for a food vacuole hemoglobinase has been identified (31) and biochemically (70-72) and molecularly (73) characterized. This protease, called falcipain, degraded denatured and native hemoglobin in vitro; its acid pH optimum, substrate specificity, and inhibitor sensitivity indicated that it was a papain family cysteine protease (64,70,71). Specific inhibitors of falcipain blocked hemoglobin degradation and prevented parasite development. The degree of inhibition of falcipain by fluoromethyl ketones (44) and vinyl sulfones (46) correlated with their inhibition of hemoglobin degradation and parasite development, supporting the hypothesis that falcipain is the cysteine protease required for hemoglobin degradation.
The specific mechanism for hemoglobin degradation in the malarial food vacuole remains unclear. As noted above, both the aspartic protease plasmepsin I and the cysteine protease falcipain have been identified in parasite food vacuoles and shown to cleave denatured and native hemoglobin in vitro (53,71). Results showing that only cysteine protease inhibitors block hemoglobin processing and globin hydrolysis in cultured parasites suggest that falcipain is required for initial steps of hemoglobin degradation (66-68,74). However, other studies have shown that native hemoglobin is cleaved by plasmepsin I, but not falcipain, in nonreducing conditions that may be present in the food vacuole (53,59,72). In any event, regardless of the exact sequence of hemoglobin processing, multiple enzymes, including at least the three proteases already identified, appear to participate in the degradation of hemoglobin. These proteases are thus logical targets for antimalarial drug development.
Aminopeptidase activity has also been described in malaria parasites (75-77). This activity, with a neutral pH optimum, was not found in food vacuole lysates (77). When these lysates were incubated with hemoglobin, discrete peptide fragments, but not free amino acids, were identified (77). These results suggest that hemoglobin is degraded to small peptides in the food vacuole, that these peptides are transported to the parasite cytosol, and that additional processing of hemoglobin peptides is mediated by cytosolic aminopeptidase activity (77).
Drug Development Efforts
Both the cysteine protease inhibitor E-64 and the aspartic protease inhibitor pepstatin blocked P. falciparum development (63-67). Administered together, the two inhibitors acted synergistically (67). However, only E-64 blocked globin hydrolysis (64-67). Numerous peptide-based cysteine protease inhibitors, including fluoromethyl ketones (44,70,78) and vinyl sulfones (46), inhibited falcipain at low nanomolar concentrations and inhibited P. falciparum development and hemoglobin degradation at concentrations below 100 nanomolar (Table 3). In a malaria animal model, a fluoromethyl ketone that inhibited falcipain at low nanomolar concentrations blocked P. vinckei protease activity in vivo after a single subcutaneous dose, and, when administered for 4 days, cured 80% of murine malaria infections (45). Thus, despite the theoretical limitations of potentially rapid degradation in vivo and inhibition of host proteases, peptide protease inhibitors show promise as candidate antimalarial drugs. Fluoromethyl ketones have subsequently shown toxicity in animal studies, but evaluations of related, apparently nontoxic inhibitors of falcipain as antimalarial drugs are under way.
A computer model for the structure of falcipain was used to identify nonpeptide inhibitors (47). Screening of potential nonpeptide inhibitors identified a low micromolar lead compound (47; Table 3). Subsequent synthesis and testing of small molecules based on the structure of the lead compound have identified biologically active falcipain inhibitors, including chalcones that block parasite metabolism at submicromolar concentrations (48) and phenothiazines that block parasite metabolism and development at low micromolar concentrations (49).
Peptidelike aspartic protease inhibitors are potent inhibitors of plasmepsins I and II. In independent studies SC-50083 (41), Ro 40-4388 (42), and "compound 7" (43) inhibited plasmepsin I or II at nanomolar concentrations and blocked parasite development at high nanomolar to micromolar concentrations (Table 3). Drug development efforts should be assisted by the recent determination of the structure of plasmepsin II (43). Inhibitors of aspartic and cysteine proteases have synergistic effects in inhibiting the growth of cultured malaria parasites (67), and these proteases also act synergistically to degrade hemoglobin in vitro (41). Therefore, the combination of inhibitors of malarial cysteine and aspartic proteases may provide the most effective chemotherapeutic regimen and best limit the development of parasite resistance to protease inhibitors. Ultimately, a better understanding of the biochemical properties and biologic roles of malarial proteases will foster the development of protease inhibitors that specifically inhibit parasite enzymes and thus are the most suitable candidates for chemotherapy.
Dr. Rosenthal is associate professor, Department of Medicine, Division of Infectious Diseases, San Francisco General Hospital and the University of California, San Francisco. His research interests include the evaluation of proteases of malaria parasites as chemotherapeutic targets.
Acknowledgment
Work in the author's laboratory was supported by grants from the National Institutes of Health, the UNDP/World Bank/WHO Special Programme for Research and Training in Tropical Diseases, and the American Heart Association.
References
- Walsh JA. Disease problems in the Third World. Ann N Y Acad Sci. 1989;569:1–16. DOIPubMedGoogle Scholar
- Oaks SC, Mitchell VS, Pearson GW, Carpenter CCJ, eds. Malaria: obstacles and opportunities. Washington: National Academy Press; 1991.
- Olliaro P, Cattani J, Wirth D. Malaria, the submerged disease. JAMA. 1996;275:230–3. DOIPubMedGoogle Scholar
- Hoffman SL, Miller LH. Perspectives on malaria vaccine development. In: Hoffman SL, editor. Malaria vaccine development. Washington: ASM Press; 1996. p. 1-13.
- Alonso PL, Lindsay SW, Armstrong JRM, Conteh M, Hill AG, David PH, The effect of insecticide-treated bed nets on mortality of Gambian children. Lancet. 1991;337:1499–502. DOIPubMedGoogle Scholar
- Collins FH, Besansky NJ. Vector biology and the control of malaria in Africa. Science. 1994;264:1874–5. DOIPubMedGoogle Scholar
- Centers for Disease Control. Recommendations for the prevention of malaria among travelers. JAMA. 1990;263:2729–40. DOIPubMedGoogle Scholar
- Murphy GS, Basri H, Purnomo , Andersen EM, Bangs MJ, Mount DL, . Vivax malaria resistant to treatment and prophylaxis with chloroquine. Lancet. 1993;341:96–100. DOIPubMedGoogle Scholar
- Wyler DJ. Malaria chemoprophylaxis for the traveler. N Engl J Med. 1993;329:31–7. DOIPubMedGoogle Scholar
- Olliaro P, Nevill C, LeBras J, Ringwald P, Mussano P, Garner P, Systematic review of amodiaquine treatment in uncomplicated malaria. Lancet. 1996;348:1196–201. DOIPubMedGoogle Scholar
- Palmer KJ, Holliday SM, Brogden RN. Mefloquine: a review of its antimalarial activity, pharmacokinetic properties and therapeutic efficacy. Drugs. 1993;45:430–75. DOIPubMedGoogle Scholar
- Luzzi GA, Peto TEA. Adverse effects of antimalarials: an update. Drug Saf. 1993;8:295–311. DOIPubMedGoogle Scholar
- Hellgren U, Kihamia CM, Bergqvist Y, Lebbad M, Premji Z, Rombo L. Standard and reduced doses of sulfadoxine-pyrimethamine for treatment of Plasmodium falciparum in Tanzania, with determination of drug concentrations and susceptibility in vitro. Trans R Soc Trop Med Hyg. 1990;84:469–73. DOIPubMedGoogle Scholar
- Bradley DJ, Warhurst DC. Malaria prophylaxis: guidelines for travellers from Britain. BMJ. 1995;310:709–14.PubMedGoogle Scholar
- Bryson HM, Goa KL. Halofantrine: a review of its antimalarial activity, pharmacokinetic properties and therapeutic potential. Drugs. 1992;43:236–58. DOIPubMedGoogle Scholar
- Meshnick SR, Taylor TE, Kamchonwongpaisan S. Artemisinin and the antimalarial endoperoxides: from herbal remedy to targeted chemotherapy. Microbiol Rev. 1996;60:301–15.PubMedGoogle Scholar
- de Vries PJ, Dien TK. Clinical pharmacology and therapeutic potential of artemisinin and its derivatives in the treatment of malaria. Drugs. 1996;52:818–36. DOIPubMedGoogle Scholar
- Radloff PD, Philipps J, Nkeyi M, Hutchinson D, Kremsner PG. Atovaquone and proguanil for Plasmodium falciparum malaria. Lancet. 1996;347:1511–4. DOIPubMedGoogle Scholar
- Ringwald P, Bickii J, Basco L. Randomised trial of pyronaridine versus chloroquine for acute uncomplicated falciparum malaria in Africa. Lancet. 1996;347:24–8. DOIPubMedGoogle Scholar
- Gordeuk VR, Thuma PE, Brittenham GM, Biemba G, Zulu S, Simwanza G, Iron chelation as a chemotherapeutic strategy for falciparum malaria. Am J Trop Med Hyg. 1993;48:193–7.PubMedGoogle Scholar
- Gordeuk V, Thuma P, Brittenham G, McLaren C, Parry D, Backenstose A, Effect of iron chelation therapy on recovery from deep coma in children with cerebral malaria. N Engl J Med. 1992;327:1473–7.PubMedGoogle Scholar
- Anderson SL, Berman J, Kuschner R, Wesche D, Magill A, Wellde B, Prophylaxis of Plasmodium falciparum malaria with azithromycin administered to volunteers. Ann Intern Med. 1995;123:771–3.PubMedGoogle Scholar
- McKerrow JH, Sun E, Rosenthal PJ, Bouvier J. The proteases and pathogenicity of parasitic protozoa. Annu Rev Microbiol. 1993;47:821–53. DOIPubMedGoogle Scholar
- Blackman MJ, Holder AA. Secondary processing of the Plasmodium falciparum merozoite surface protein-1 (MSP1) by a calcium-dependent membrane-bound serine protease: shedding of MSP133 as a noncovalently associated complex with other fragments of the MSP1. Mol Biochem Parasitol. 1992;50:307–16. DOIPubMedGoogle Scholar
- Perkins ME. Erythrocyte invasion by the malarial merozoite: recent advances. Exp Parasitol. 1989;69:94–9. DOIPubMedGoogle Scholar
- Bernard F, Schrevel J. Purification of a Plasmodium berghei neutral endopeptidase and its localization in merozoite. Mol Biochem Parasitol. 1987;26:167–74. DOIPubMedGoogle Scholar
- Rosenthal PJ, Kim K, McKerrow JH, Leech JH. Identification of three stage-specific proteinases of Plasmodium falciparum. J Exp Med. 1987;166:816–21. DOIPubMedGoogle Scholar
- Braun-Breton C, Rosenberry TL, Pereira da Silva L. Induction of the proteolytic activity of a membrane protein in Plasmodium falciparum by phosphatidyl inositol-specific phospholipase C. Nature. 1988;332:457–9. DOIPubMedGoogle Scholar
- Roggwiller E, Bétoulle MEM, Blisnick T, Braun Breton C. A role for erythrocyte band 3 degradation by the parasite gp76 serine protease in the formation of the parasitophorous vacuole during invasion of erythrocytes by Plasmodium falciparum. Mol Biochem Parasitol. 1996;82:13–24. DOIPubMedGoogle Scholar
- Deguercy A, Hommel M, Schrevel J. Purification and characterization of 37-kilodalton proteases from Plasmodium falciparum and Plasmodium berghei which cleave erythrocyte cytoskeletal components. Mol Biochem Parasitol. 1990;38:233–44. DOIPubMedGoogle Scholar
- Knapp B, Hundt E, Nau U, Küpper HA. Molecular cloning, genomic structure and localization in a blood stage antigen of Plasmodium falciparum characterized by a serine stretch. Mol Biochem Parasitol. 1989;32:73–84. DOIPubMedGoogle Scholar
- Li W-B, Bzik DJ, Horii T, Inselburg J. Structure and expression of the Plasmodium falciparum SERA gene. Mol Biochem Parasitol. 1989;33:13–26. DOIPubMedGoogle Scholar
- Knapp B, Nau U, Hundt E, Küpper HA. A new blood stage antigen of Plasmodium falciparum highly homologous to the serine-stretch protein SERP. Mol Biochem Parasitol. 1991;44:1–14. DOIPubMedGoogle Scholar
- Roggwiller E, Fricaud A-C, Blisnick T, Braun-Breton C. Host urokinase-type plasminogen activator participates in the release of malaria merozoites from infected erythrocytes. Mol Biochem Parasitol. 1997;86:49–59. DOIPubMedGoogle Scholar
- Schrevel J, Grellier P, Mayer R, Monsigny M. Neutral proteases involved in the reinvasion of erythrocytes by Plasmodium merozoites. Biol Cell. 1988;64:233–44. DOIPubMedGoogle Scholar
- Mayer R, Picard I, Lawton P, Grellier P, Barrault C, Monsigny M, Peptide derivatives specific for a Plasmodium falciparum proteinase inhibit the human erythrocyte invasion by merozoites. J Med Chem. 1991;34:3029–35. DOIPubMedGoogle Scholar
- Francis SE, Gluzman IY, Oksman A, Knickerbocker A, Mueller R, Bryant ML, Molecular characterization and inhibition of a Plasmodium falciparum aspartic hemoglobinase. EMBO J. 1994;13:306–17.PubMedGoogle Scholar
- Moon RP, Tyas L, Certa U, Rupp K, Bur D, Jacquet C, Expression and characterisation of plasmepsin I from Plasmodium falciparum. Eur J Biochem. 1997;244:552–60. DOIPubMedGoogle Scholar
- Silva AM, Lee AY, Gulnik SV, Majer P, Collins J, Bhat TN, Structure and inhibition of plasmepsin II, a hemoglobin-degrading enzyme from Plasmodium falciparum. Proc Natl Acad Sci U S A. 1996;93:10034–9. DOIPubMedGoogle Scholar
- Rosenthal PJ, Wollish WS, Palmer JT, Rasnick D. Antimalarial effects of peptide inhibitors of a Plasmodium falciparum cysteine proteinase. J Clin Invest. 1991;88:1467–72. DOIPubMedGoogle Scholar
- Rosenthal PJ, Lee GK, Smith RE. Inhibition of a Plasmodium vinckei cysteine proteinase cures murine malaria. J Clin Invest. 1993;91:1052–6. DOIPubMedGoogle Scholar
- Rosenthal PJ, Olson JE, Lee GK, Palmer JT, Klaus JL, Rasnick D. Antimalarial effects of vinyl sulfone cysteine proteinase inhibitors. Antimicrob Agents Chemother. 1996;40:1600–3.PubMedGoogle Scholar
- Ring CS, Sun E, McKerrow JH, Lee GK, Rosenthal PJ, Kuntz ID, Structure-based inhibitor design by using protein models for the development of antiparasitic agents. Proc Natl Acad Sci U S A. 1993;90:3583–7. DOIPubMedGoogle Scholar
- Li R, Kenyon GL, Cohen FE, Chen X, Gong B, Dominguez JN, In vitro antimalarial activity of chalcones and their derivatives. J Med Chem. 1995;38:5031–7. DOIPubMedGoogle Scholar
- Domínguez JN, López S, Charris J, Iarruso L, Lobo G, Semenov A, Synthesis and antimalarial effects of phenothiazine inhibitors of a Plasmodium falciparum cysteine protease. J Med Chem. 1997;40:2726–32. DOIPubMedGoogle Scholar
- Rosenthal PJ, Meshnick SR. Hemoglobin catabolism and iron utilization by malaria parasites. Mol Biochem Parasitol. 1996;83:131–9. DOIPubMedGoogle Scholar
- Bond JS, Butler PE. Intracellular proteases. Annu Rev Biochem. 1987;56:333–64. DOIPubMedGoogle Scholar
- Gluzman IY, Francis SE, Oksman A, Smith CE, Duffin KL, Goldberg DE. Order and specificity of the Plasmodium falciparum hemoglobin degradation pathway. J Clin Invest. 1994;93:1602–8. DOIPubMedGoogle Scholar
- Gyang FN, Poole B, Trager W. Peptidases from Plasmodium falciparum cultured in vitro. Mol Biochem Parasitol. 1982;5:263–73. DOIPubMedGoogle Scholar
- Aissi E, Charet P, Bouquelet S, Biguet J. Endoprotease in Plasmodium yoelii nigeriensis. Comp Biochem Physiol. 1983;74B:559–66.
- Sherman IW, Tanigoshi L. Purification of Plasmodium lophurae cathepsin D and its effects on erythrocyte membrane proteins. Mol Biochem Parasitol. 1983;8:207–26. DOIPubMedGoogle Scholar
- Sato K, Fukabori Y, Suzuki M. Plasmodium berghei: a study of globinolytic enzyme in erythrocytic parasite. Zbl Bakt Hyg A. 1987;264:487–95.
- Bailly E, Savel J, Mahouy G, Jaureguiberry G. Plasmodium falciparum: isolation and characterization of a 55-kDa protease with a cathepsin D-like activity from P. falciparum. Exp Parasitol. 1991;72:278–84. DOIPubMedGoogle Scholar
- Goldberg DE, Slater AFG, Beavis R, Chait B, Cerami A, Henderson GB. Hemoglobin degradation in the human malaria pathogen Plasmodium falciparum: a catabolic pathway initiated by a specific aspartic protease. J Exp Med. 1991;173:961–9. DOIPubMedGoogle Scholar
- Vander Jagt DL, Hunsaker LA, Campos NM, Scaletti JV. Localization and characterization of hemoglobin-degrading aspartic proteinases from the malarial parasite Plasmodium falciparum. Biochim Biophys Acta. 1992;1122:256–64.PubMedGoogle Scholar
- Dame JB, Reddy GR, Yowell CA, Dunn BM, Kay J, Berry C. Sequence, expression and modeled structure of an aspartic proteinase from the human malaria parasite Plasmodium falciparum. Mol Biochem Parasitol. 1994;64:177–90. DOIPubMedGoogle Scholar
- Hill J, Tyas L, Phylip LH, Kay J, Dunn BM, Berry C. High level expression and characterisation of plasmepsin II, an aspartic proteinase from Plasmodium falciparum. FEBS Lett. 1994;352:155–8. DOIPubMedGoogle Scholar
- Dluzewski AR, Rangachari K, Wilson RJM, Gratzer WB. Plasmodium falciparum: protease inhibitors and inhibition of erythrocyte invasion. Exp Parasitol. 1986;62:416–22. DOIPubMedGoogle Scholar
- Rosenthal PJ, McKerrow JH, Aikawa M, Nagasawa H, Leech JH. A malarial cysteine proteinase is necessary for hemoglobin degradation by Plasmodium falciparum. J Clin Invest. 1988;82:1560–6. DOIPubMedGoogle Scholar
- Vander Jagt DL, Caughey WS, Campos NM, Hunsaker LA, Zanner MA. Parasite proteases and antimalarial activities of protease inhibitors. Prog Clin Biol Res. 1989;313:105–18.PubMedGoogle Scholar
- Rosenthal PJ. Plasmodium falciparum: effects of proteinase inhibitors on globin hydrolysis by cultured malaria parasites. Exp Parasitol. 1995;80:272–81. DOIPubMedGoogle Scholar
- Bailly E, Jambou R, Savel J, Jaureguiberry G. Plasmodium falciparum: differential sensitivity in vitro to E-64 (cysteine protease inhibitor) and pepstatin A (aspartyl protease inhibitor). J Protozool. 1992;39:593–9.PubMedGoogle Scholar
- Gamboa de Domínguez ND, Rosenthal PJ. Cysteine proteinase inhibitors block early steps in hemoglobin degradation by cultured malaria parasites. Blood. 1996;87:4448–54.PubMedGoogle Scholar
- Asawamahasakda W, Ittarat I, Chang C-C, McElroy P, Meshnick SR. Effects of antimalarials and protease inhibitors on plasmodial hemozoin production. Mol Biochem Parasitol. 1994;67:183–91. DOIPubMedGoogle Scholar
- Rosenthal PJ, McKerrow JH, Rasnick D, Leech JH. Plasmodium falciparum: inhibitors of lysosomal cysteine proteinases inhibit a trophozoite proteinase and block parasite development. Mol Biochem Parasitol. 1989;35:177–84. DOIPubMedGoogle Scholar
- Salas F, Fichmann J, Lee GK, Scott MD, Rosenthal PJ. Functional expression of falcipain, a Plasmodium falciparum cysteine proteinase, supports its role as a malarial hemoglobinase. Infect Immun. 1995;63:2120–5.PubMedGoogle Scholar
- Francis SE, Gluzman IY, Oksman A, Banerjee D, Goldberg DE. Characterization of native falcipain, an enzyme involved in Plasmodium falciparum hemoglobin degradation. Mol Biochem Parasitol. 1996;83:189–200. DOIPubMedGoogle Scholar
- Rosenthal PJ, Nelson RG. Isolation and characterization of a cysteine proteinase gene of Plasmodium falciparum. Mol Biochem Parasitol. 1992;51:143–52. DOIPubMedGoogle Scholar
- Kamchonwongpaisan S, Samoff E, Meshnick SR. Identification of hemoglobin degradation products in Plasmodium falciparum. Mol Biochem Parasitol. 1997;86:179–86. DOIPubMedGoogle Scholar
- Vander Jagt DL, Baack BR, Hunsaker LA. Purification and characterization of an aminopeptidase from Plasmodium falciparum. Mol Biochem Parasitol. 1984;10:45–54. DOIPubMedGoogle Scholar
- Curley GP, O'Donovan SM, McNally J, Mullally M, O'Hara H, Troy A, Aminopeptidases from Plasmodium falciparum, Plasmodium chabaudi, and Plasmodium berghei. J Eukaryot Microbiol. 1994;41:119–23. DOIPubMedGoogle Scholar
- Kolakovich KA, Gluzman IY, Duffin KL, Goldberg DE. Generation of hemoglobin peptides in the acidic digestive vacuole of Plasmodium falciparum implicates peptide transport in amino acid production. Mol Biochem Parasitol. 1997;87:123–35. DOIPubMedGoogle Scholar
- Rockett KA, Playfair JHL, Ashall F, Targett GAT, Angliker H, Shaw E. Inhibition of intraerythrocytic development of Plasmodium falciparum by proteinase inhibitors. FEBS Lett. 1990;259:257–9. DOIPubMedGoogle Scholar
Figure
Tables
Cite This ArticleTable of Contents – Volume 4, Number 1—March 1998
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Philip Rosenthal, Box 0811, University of California, San Francisco, CA 94143-0811, USA; fax: 415-206-6015
Top