Volume 4, Number 2—June 1998
Synopsis
Multiple-Drug Resistant Enterococci: The Nature of the Problem and an Agenda for the Future
Cite This Article
Citation for Media
Abstract
Enterococci, leading causes of nosocomial bacteremia, surgical wound infection, and urinary tract infection, are becoming resistant to many and sometimes all standard therapies. New rapid surveillance methods are highlighting the importance of examining enterococcal isolates at the species level. Most enterococcal infections are caused by Enterococcus faecalis, which are more likely to express traits related to overt virulence but—for the moment—also more likely to retain sensitivity to at least one effective antibiotic. The remaining infections are mostly caused by E. faecium, a species virtually devoid of known overt pathogenic traits but more likely to be resistant to even antibiotics of last resort. Effective control of multiple-drug resistant enterococci will require 1) better understanding of the interaction between enterococci, the hospital environment, and humans, 2) prudent antibiotic use, 3) better contact isolation in hospitals and other patient care environments, and 4) improved surveillance. Equally important is renewed vigor in the search for additional drugs, accompanied by the evolution of new therapeutic paradigms less vulnerable to the cycle of drug introduction and drug resistance.
The past few years have witnessed increasing interest in enterococci. Until recently, these ordinary bowel commensals languished as misclassified streptococci, commonly perceived "with the exception of endocarditis and rare cases of meningitis … [as] not … a major cause of serious infection" (1). In the last decade, however, enterococci have become recognized as leading causes of nosocomial bacteremia, surgical wound infection, and urinary tract infection (2,3). Two types of enterococci cause infections: 1) those originating from patients' native flora, which are unlikely to possess resistance beyond that intrinsic to the genus and are unlikely to be spread from bed to bed, and 2) isolates that possess multiple antibiotic resistance traits and are capable of nosocomial transmission. The therapeutic challenge of multiple-drug resistant (MDR) enterococci—those strains with significant resistance to two or more antibiotics, often including, but not limited to, vancomycin—has brought their role as important nosocomial pathogens into sharper focus.
The accretion and spread of antibiotic resistance determinants among enterococci, to the point where some clinical isolates are resistant to all standard therapies, highlight both the vulnerability of our present armament as well as the looming prospect of a "postantibiotic era" (4). This review focuses on the magnitude and nature of the problem posed by enterococci in general, and MDR enterococci in particular. For many points, only representative citations are provided.
Enterococci normally inhabit the bowel. They are found in the intestine of nearly all animals, from cockroaches to humans. Enterococci are readily recovered outdoors from vegetation and surface water, probably because of contamination by animal excrement or untreated sewage (5). In humans, typical concentrations of enterococci in stool are up to 108 CFU per gram (6). Although the oral cavity and vaginal tract can become colonized, enterococci are recovered from these sites in fewer than 20% of cases. The predominant species inhabiting the intestine varies. In Europe, the United States, and the Far East, Enterococcus faecalis predominates in some instances and E. faecium in others (6). Ecologic or microbial factors promoting intestinal colonization are obscure. Of 14 or more enterococcal species (7), only E. faecalis and E. faecium commonly colonize and infect humans in detectable numbers. E. faecalis is isolated from approximately 80% of human infections, and E. faecium from most of the rest. Infections to other enterococcal species are rare.
Enterococci are exceedingly hardy. They tolerate a wide variety of growth conditions, including temperatures of 10°C to 45°C, and hypotonic, hypertonic, acidic, or alkaline environments. Sodium azide and concentrated bile salts, which inhibit or kill most microorganisms, are tolerated by enterococci and used as selective agents in agar-based media. As facultative organisms, enterococci grow under reduced or oxygenated conditions. Enterococci are usually considered strict fermenters because they lack a Kreb's cycle and respiratory chain (8). E. faecalis is an exception since exogenous hemin can be used to produce d, b, and o type cytochromes (9,10). In a survey of 134 enterococci and related streptococci, only E. faecalis and Lactococcus lactis expressed cytochrome-like respiration (11). Cytochromes provide a growth advantage to E. faecalis during aerobic growth (9). E. faecalis cytochromes are only expressed under aerobic conditions in the presence of exogenous hemin (9,10,12) and, therefore, may promote the colonization of inappropriate sites.
Enterococci are intrinsically resistant to many antibiotics. Unlike acquired resistance and virulence traits, which are usually transposon or plasmid encoded, intrinsic resistance is based in chromosomal genes, which typically are nontransferrable. Penicillin, ampicillin, piperacillin, imipenem, and vancomycin are among the few antibiotics that show consistent inhibitory, but not bactericidal, activity against E. faecalis. E. faecium are less susceptible to ß-lactam antibiotics than E. faecalis because the penicillin-binding proteins of the former have markedly lower affinities for the antibiotics (13). The first reports of strains highly resistant to penicillin began to appear in the 1980s (14,15).
Enterococci often acquire antibiotic resistance through exchange of resistance-encoding genes carried on conjugative transposons, pheromone-responsive plasmids, and other broad-host-range plasmids (6). The past two decades have witnessed the rapid emergence of MDR enterococci. High-level gentamicin resistance occurred in 1979 (16) and was quickly followed by numerous reports of nosocomial infection in the 1980s (17). Simultaneously, sporadic outbreaks of nosocomial E. faecalis and E. faecium infection appeared with penicillin resistance due to ß-lactamase production (18); however, such isolates remain rare. Finally, MDR enterococci that had lost susceptibility to vancomycin were reported in Europe (19,20) and the United States (21).
Among several phenotypes for vancomycin-resistant enterococci, VanA (resistance to vancomycin and teicoplanin) and VanB (resistance to vancomycin alone) are most common (22). In the United States, VanA and VanB account for approximately 60% and 40% of vancomycin-resistant enterococci (VRE) isolates, respectively (23). Inducible genes encoding these phenotypes alter cell wall synthesis and render strains resistant to glycopeptides (22).
VanA and VanB types of resistance are primarily found among enterococci isolated from clinical, veterinary, and food specimens (24), but not other common intestinal or environmental bacteria. In the laboratory, however, these genes can be transferred from enterococci to other bacteria (22). For example, Staphylococcus aureus has been rendered vancomycin-resistant through apparent transfer of resistance from E. faecalis on the surface of membrane filters and on the skin of hairless obese mice (25), which indicates that there is no biologic barrier to the emergence of vancomycin-resistant S. aureus. Clinical isolates of highly vancomycin-resistant S. aureus have yet to be identified, although strains with reduced susceptibility to vancomycin have appeared (26). The mechanism of resistance for these strains remains undetermined but does not appear to involve genes associated with VanA or VanB phenotypes.
Enterococci account for approximately 110,000 urinary tract infections, 25,000 cases of bacteremia, 40,000 wound infections, and 1,100 cases of endocarditis annually in the United States (2,27,28). Most infections occur in hospitals. Although several studies have suggested an increase in nosocomial infection rates for enterococci in recent years, National Nosocomial Infections Surveillance system data show little change in the percentage of enterococcal bloodstream (12% vs. 7%), surgical site (15% vs. 11%), and urinary tract (14% vs. 14%) infections over the past 2 decades (3,29). Adequate surveillance data prior to 1980 are not available. Enterococcal infection deaths have also been difficult to ascertain because severe comorbid illnesses are common; however, enterococcal sepsis is implicated in 7% to 50% of fatal cases (6). Several case-control and historical cohort studies show that death risk associated with antibiotic-resistant enterococcal bacteremia is severalfold higher than death risk associated with susceptible enterococcal bacteremia (30). This trend will likely increase as MDR isolates become more prevalent.
Colonization and infection with MDR enterococci occur worldwide. Early reports showed that in the United States, the percentage of nosocomial infections caused by VRE increased more than 20-fold (from 0.3% to 7.9%) between 1989 and 1993, indicating rapid dissemination. New database technologies, such as The Surveillance Network (TSN) Database-USA, now permit the assessment of resistance profiles according to species. TSN Database electronically collects and compiles data daily from more than 100 U.S. clinical laboratories, identifies potential laboratory testing errors, and detects emergence of resistance profiles and mechanisms that pose a public health threat (e.g., vancomycin-resistant staphylococci).
Figure 1

Figure 1. Epidemiology of enterococcal infection based on 15,203 susceptibility results obtained by The Surveillance Network (TSN) Database-USA, 1995 to Sep 1, 1997. The increase in total numbers between 1995 and 1996 represents...
Data collected by the TSN Database between 1995 and September 1, 1997 were analyzed to determine whether the earlier increase in vancomycin resistance was unique to vancomycin, whether it represented a continuing trend, and whether speciation is quantifiably important in analyzing this trend. E. faecalis resistance to ampicillin and vancomycin is uncommon (Figure 1); little change in resistance prevalence occurred from 1995 to 1997. In contrast, E. faecium vancomycin and ampicillin resistance increased alarmingly. In 1997, 771 (52%) of 1,482 of E. faecium isolates exhibited vancomycin resistance, and 1,220 (83%) of 1,474 isolates exhibited ampicillin resistance (Figure 1). E. faecium resistance notwithstanding, E. faecalis remained by far the most commonly encountered of the two enterococcal species in TSN Database. E. faecalis to E. faecium total isolates were approximately 4:1 (Figure 1), blood isolates 3:1, and urine isolates 5:1. This observation underscores important differences in the survival strategies and likelihood of therapeutic success, critical factors usually obscured by lumping the organisms together as Enterococcus species or enterococci. Widespread emergence and dissemination of ampicillin and vancomycin resistance in E. faecalis would significantly confound the current therapeutic dilemma. There is little reason to suspect that vancomycin and ampicillin resistances only provide selective advantage for the species faecium and not faecalis. The relative absence of these resistances in E. faecalis may simply reflect a momentary lack of penetrance and equilibration of the traits. Because of these important differences between the two species, meaningful surveillance of enterococcal resistance must include species identification.
Although exact modes of nosocomial transmission for MDR enterococci are difficult to prove, molecular microbiologic and epidemiologic evidence strongly suggest spread between patients, probably on the hands of health-care providers or medical devices, and between hospitals by patients with prolonged intestinal colonization. At least 16 outbreaks of MDR enterococci have been reported since 1989 (31); all but two were due to E. faecium. This disparity, particularly in view of the higher numbers of clinical E. faecalis isolates, may reflect a reporting bias due to the novelty of the combinations of resistance that occur in E. faecium. When isolates from outbreaks of MDR enterococci have been analyzed by genetic fingerprints, more than half involve clonally related isolates (18,32).
Prior treatment with antibiotics is common in nearly all patients colonized or infected with MDR enterococci (33-35). Clindamycin, cephalosporin, aztreonam, ciprofloxacin, aminoglycoside, and metronidazole use is equally or more often associated with colonization or infection with MDR enterococci than vancomycin use. Other risk factors include prolonged hospitalization; high severity of illness score; intraabdominal surgery; renal insufficiency; enteral tube feedings; and exposure to specific hospital units, nurses, or contaminated objects and surfaces within patient-care areas.
Controlling the spread of MDR enterococci among inpatients is difficult. We know relatively little about the biology of enterococcal transmission or the specific microbial factors favoring colonization by exogenous enterococcal strains. Nevertheless, VRE infection control policies, which could apply to MDR enterococci, were recently published by the Hospital Infection Control Practices Advisory Committee (36). Control methods include routine screening for vancomycin resistance among clinical isolates, active surveillance for VRE in intensive care units, contact isolation to minimize person-to-person transmission, and vancomycin restriction.
These measures to limit VRE spread, however, have failed on occasion (35). Not all hospitals can or are willing to perform active surveillance. Because more patients are typically colonized with VRE (3% to 47%) than are infected (35,37,38), and because intestinal colonization can be prolonged, passive surveillance by routine cultures allows colonized inpatients to go unidentified and serve as point sources for continued spread of VRE. Even if all colonized inpatients are successfully identified, VRE may be spread by health-care workers through either inadequate hand washing (39) or through contact with items such as bedrails, sinks, faucets, and doorknobs (enterococci can persist for weeks on environmental surfaces) (40). Decontamination efforts must be rigorous.
The Hospital Infection Control Practices Advisory Committee strongly recommended restricting oral and parenteral vancomycin to control VRE (36). However, limiting use of vancomycin while ignoring widespread use of other broad spectrum antibiotics likely will not lead to maximal control of VRE or of MDR enterococci.
Antibiotics may promote colonization and infection with MDR enterococci by at least two mechanisms. First, many broad spectrum antibiotics have little or no anti-enterococcal activity, and administration commonly leads to overgrowth of susceptible (or resistant) enterococci at sites at risk for infection. Second, most antibiotics substantially reduce the normal resistance of the intestinal tract to colonization by exogenous organisms (41). Colonization resistance results primarily from the "limiting action" of the normal anaerobic flora, and to a lesser extent from an intact mucosa, gastric acid secretion, intestinal motility, and intestinal-associated immunity (41). Antibiotic-induced alterations in the protective flora of the intestine provide large footholds for colonization with exogenous pathogens such as MDR enterococci (41). Antibiotic restriction programs would be more effective if they included prudent prescribing of all antibiotics, not just single agents such as vancomycin. This approach substantially decreased intestinal colonization with VRE in one hospital pharmacy that restricted vancomycin, cefotaxime, and clindamycin (42).
At a minimum, a successful program for control of MDR enterococci requires effective passive and active surveillance to identify colonized and infected patients, absolute adherence to contact isolation by health-care workers, rigorous decontamination of patient-contact areas and judicious use or restriction of vancomycin and other broad spectrum antibiotics.
Suitable antibiotics often are not available to treat MDR enterococcal infections, e.g., endocarditis or bacteremia, in the presence of neutropenia. Combinations of penicillin with vancomycin, ciprofloxacin with ampicillin, or novobiocin with doxycycline, among others, have been used (43) but can be unpredictable and remain clinically unproven. In one report chloramphenicol successfully treated chloramphenicol-susceptible infections (44), but these findings await confirmation in controlled trials.
Promising new antibiotics for MDR enterococcal infection under investigation include fluoroquinolones, streptogramins, oxazolidinones, semisynthetic glycopeptides, and glycylcyclines. Clinafloxacin, a fluoroquinolone with improved potency against enterococci compared with ciprofloxacin, has excellent activity against VRE, appears bactericidal in vitro, and has been effective in treatment of enterococcal infections in a murine model (45). Although single-step resistance to clinafloxacin could not be detected in vitro, multistep resistance is readily achieved. Should this agent gain approval for treatment of enterococcal infections, selection for resistance may limit its effectiveness.
Quinupristin/dalfopristin (Synercid) is a combination of streptogramins A and B that inhibits protein synthesis and has a narrower spectrum of activity against enterococci than clinafloxacin (46). Many, but not all, E. faecium isolates with VanA and VanB phenotypes are susceptible (47); however, E. faecalis is uniformly resistant, and superinfection has been reported during therapy (48). In addition, quinupristin/dalfopristin is bacteriostatic only, potentially allowing emergence of resistance (49). For these reasons the drug may have only a limited role in treating MDR enterococcal infections. Novel oxazolidinones and glycylcyclines have also shown potent activity against enterococci, including MDR enterococci (50,51), but await further testing.
The substantial drawback of the broad spectrum approach is that the more organisms affected (both protective commensals as well as pathogens), the more opportunities for resistance to evolve. Broad spectrum antibiotics permit empiric therapy in the absence of a specific diagnosis and generate a more substantial return on investment in the short term. However, broad spectrum antibiotics affect not only disease-causing organisms but also commensals present in numbers large enough to generate resistance by otherwise rare mutations or genetic exchange events. As long as market forces favor development of broad spectrum therapeutics, a cycle of drug introduction followed by emergence of resistance undoubtedly will continue.
In contrast to the historical reliance on broad spectrum antibiotic therapy, the continuing development and introduction of rapid diagnostic techniques (52) may allow a more focused approach to infectious disease therapy. Any of the myriad microbial-host interactions that subvert the host response or damage tissues during an infection represent potential therapeutic targets. However, many key interactions in disease pathogenesis are specific to the organism involve—da characteristic that is both a strength and a weakness. Because of the specificity of these interactions, rapid and accurate diagnosis is required. However, therapeutics aimed only at interaction between host and a specific pathogen should leave the diverse commensal flora essentially unaffected. As a result, the targeted population would be restricted to the relatively small numbers of disease-producing bacteria and would not likely reach the numbers or diversity required to make development of resistance a statistical probability.
The current spectrum of approaches to identify new antiinfective compounds has two extremes: 1) screening vast libraries of compounds to identify substances that by chance inhibit a microbial property and 2) detailed study of interactions between host and parasite to identify critical events leading to host tissue damage or compromise (53).
With a long-term view toward new therapeutic approaches as well as optimal use of existing therapies, we and others have begun examining in detail the interactions between enterococci and host (6). A major obstacle is that enterococci also form part of the commensal or autochthonous flora; as such, they are nearly devoid of traits traditionally associated with overt pathogens and have subtle interactions with the host. Using inocula with as few as 10 organisms, we have developed sensitive biologic systems for examining the host-parasite interactions (54).
Figure 2
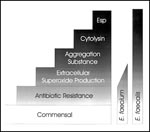
Figure 2. Virulence traits and their association with enterococcal species.
Although E. faecium strains are resistant to vancomycin and ampicillin more often than E. faecalis strains, the relative proportion of infections caused by these species has not dramatically changed in recent years (Figure 1). Since both organisms are frequently isolated from the commensal flora, this bias suggests that E. faecalis traits confer a greater degree of intrinsic virulence, for example, cytolysin production, pheromone-responsive plasmid transfer (and accompanying production of aggregation substance), extracellular superoxide production, and a newly identified surface protein tentatively termed Esp (5,56,57) (Figure 2). These properties provide logical points of departure for developing new targeted therapeutic approaches to enterococcal disease; examination of more subtle interactions between E. faecium and host will follow as an understanding of enterococcal biology evolves.
Targeting the E. faecalis Cytolysin
Figure 3

Cytolysin is disproportionately expressed by E. faecalis strains associated with disease (5,55,56). This cytolysin causes rupture of a variety of target membranes, including bacterial cells, erythrocytes, and other mammalian cells, with activity observed as a hemolytic zone on some types of blood agar. Cytolysin contributes to the toxicity or lethality of infection in several infection models and is associated with a fivefold increased risk of sudden death from nosocomial bacteremia (54,56-59). Cytolysin also contributes to the appearance of enterococci in a murine bacteremia model (Figure 3; 45,60), an observation consistent with the disproportionate representation of cytolytic strains among human blood isolates (56,62).
Figure 4
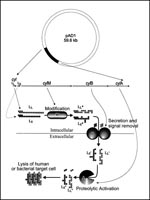
Beginning with E.W. Todd in 1934 (63) and culminating in a recent study (64), the E. faecalis cytolysin has now been characterized as a unique, extensively modified bacterial toxin (Figure 4). The cytolysin maturation pathway is ideally designed for therapeutic targeting because the two structural subunits are activated by an extracellular protease, an event that is accessible and potentially inhibitable by a novel therapeutic. Moreover, the activator protease, CylA, belongs to the subtilisin class of serine proteases (64), whose structure-function relationships and inhibitor design we are beginning to understand. Investigations are in progress to design and test inhibitors of extracellular cytolysin activation to determine whether a reduction by several logs in the levels of circulating enterococci can be attained as would be predicted by the observed behavior of cytolysin mutants (Figure 3).
An inhibitor of cytolysin activation, accompanied by appropriate rapid diagnostics, would be of potential value in treating bacteremia caused by cytolytic strains of E. faecalis without affecting commensal flora. Development of resistance should be exceedingly improbable because of the small number of bacteria targeted and because unlike antibiotics, cytolysin inhibitors would not act directly on the organism itself.
Other Enterococcal Targets
Several laboratories are using information on the E. faecalis genome and genomes of other pathogens to identify therapeutic targets (66) and facilitate studies on pathogenesis for the remaining 60% of noncytolytic enterococcal infections. The genome of an E. faecalis strain that caused multiple hospital infections (56) was sampled at high frequency by sequence analysis. Several sequences appeared to have a role in host-parasite interaction. The gene specifying Esp encodes an apparent surface protein of unusual repeating structure (67). Although a role for this protein in enterococcal infection has yet to be determined, its distribution among clinical and commensal strains is tantalizing: 29 of 30 strains with this gene were recovered from patients with bacteremia or endocarditis; one of 34 isolates obtained from healthy volunteers possessed Esp. The core of this large protein (inferred mass of 202 kDa) consists of a series of 82 amino acid repeats encoded by highly conserved tandem 246 base pair repeats. Lack of divergence in repeat sequences suggests that recombination occurs at high frequency, perhaps during infection. Moreover, the number of repeats observed in homologous genes from different E. faecalis isolates is 3 to 9 (67). This gene is flanked by a sequence similar to the transposase of IS905. None of 24 clinical or laboratory E. faecium isolates had this gene (67; V. Shankar, G. Lindahl, and M. Gilmore, unpub. data).
A second promising lead involves a series of genes encoding products highly related to enzymes involved in O-antigen synthesis in gram-negative bacteria (68). Preliminary evidence suggests that in E. faecalis these genes contribute to cell wall carbohydrate synthesis and that this carbohydrate relates to persistence in vivo. A knockout in one of these genes results in a strain with normal in vitro growth, but after subcutaneous injection, the mutant was more readily cleared than the wild type parental strain (68). One of the genes studied was present in all E. faecalis strains examined, whereas another occurs only in E. faecalis strains that share a periodate-susceptible epitope (68). Collectively, these data indicate that enzymes for synthesis of E. faecalis surface carbohydrates are important for persistence in vivo and may represent a useful therapeutic target. Taking a different approach, Arduino et al. (69,70) identified a protease-resistant, periodate susceptible substance associated with some strains of E. faecium, but not E. faecalis, which conferred resistane to phagocytosis in vitro. The relationship between the putative carbohydrate of E. faecalis under study above and the inhibitory substance of E. faecium remains to be determined. It may be found that many enterococci produce such carbohydrates at biologically significant levels in vivo, but only some strains of E. faecium do so in vitro.
Finally, recent observations indicate that nearly all E. faecalis strains, and only a few E. faecium strains, generate substantial extracellular superoxide. When E. faecalis isolates from patients with endocarditis and bacteremia were compared with isolates from healthy volunteers (71), on average, extracellular superoxide production was 60% higher among blood isolates than commensal strains. These data raised several questions: Do E. faecalis that produce larger amounts of extracellular superoxide possess greater metabolic flexibility, facilitating adaptation to nonintestinal infection sites? Does free radical production lead to host cell damage, allowing release of normally sequestered nutrients (e.g., hemin) that might promote enhanced E. faecalis growth through cytochrome formation? Might antioxidants modulate colonization or invasive infection? Answers to these questions may provide new insights into the transition from intestinal colonization to infection and may suggest new preventive strategies.
Obstacles to Further Development
Although important insights into enterococcal biology and pathogenesis are being gleaned from a reverse genetic approach, a paucity of information still exists on how enterococci colonize the intestinal tract and cause infection. For example, do E. faecalis or E. faecium colonize the colon through specific interactions with ligands on human epithelial cells or intestinal mucin? Do MDR enterococci possess alternate binding activities that enable them to colonize the intestinal tract at new sites without competing with the indigenous enterococci? Do probiotics have a role in restoring colonization resistance to an intestinal ecology altered by broad spectrum antibiotics?
Is enough being done to combat the emergence of highly resistant nosocomial pathogens? To effectively compete, industry remains highly responsive to market opportunities. Research in the public sector has been slow to respond, and as a result, our understanding of the biology of enterococcal infection is inadequate. Reasons for the modest public sector response include the following. 1) The emergence of resistant enterococci coincided with a reduction of public support for non-AIDS related infectious disease research. 2) The pathogenesis of nosocomial infection deviates from paradigms established for obligate pathogens. 3) The research infrastructure is relatively small because of the low importance traditionally attached to enterococci as etiologic agents of human disease and the deemphasis on antibiotic resistance research in the 1980s.
Historically, substantial resources have been invested in developing an in-depth understanding of the molecular biology of model organisms. During the 1960s and 1970s, when gram-negative organisms were leading causes of hospital- and community-acquired infections and gram-positive organisms were typically sensitive to existing antibiotics (72), a sizable investment in gram-negative model organisms was appropriate. However, with the emergence of gram-positive organisms as leading causes of both hospital- and community-acquired infection in the 1990s, a reevaluation of public research priorities is warranted.
Since antibiotic use became widespread 50 years ago, bacteria have steadily and routinely developed resistance. Control of the emergence of resistance will depend on new approaches to prudent antibiotic use in hospitals and clinics, based in part on improved surveillance for MDR enterococci and on better systems to encourage staff adherence to contact isolation procedures. Equally important will be development of new drugs with narrower spectra of activity aimed at known and potentially new targets and the evolution of market conditions that favor their use.
Portions of the work described were supported by Veterans Administration Merit Review Program, grants from the Public Health Service (EY08289 and AI41108), and an unrestricted award from Research to Prevent Blindness, Inc.
Dr. Huycke is an associate professor in the Infectious Diseases Section, Department of Medicine, Oklahoma University Health Sciences Center. He is interested in enterococcal pathogenesis as it relates to extracellular superoxide production by E. faecalis.
References
- Kaye D. Enterococci: biologic and epidemiologic characteristics and in vitro susceptibility. Arch Intern Med. 1982;142:2006–9. DOIPubMedGoogle Scholar
- Emori TG, Gaynes RP. An overview of nosocomial infections, including the role of the microbiology laboratory. Clin Microbiol Rev. 1993;6:428–42.PubMedGoogle Scholar
- Jarvis WR, Gaynes RP, Horan TC, Emori TG, Stroud LA, Archibald LK, Semiannual report: aggregated data from the National Nosocomial Infections Surveillance (NNIS) system. CDC, 1996:1-27.
- Cohen ML. Epidemiology of drug resistance: implications for a post-antimicrobial era. Science. 1992;257:1050–5. DOIPubMedGoogle Scholar
- Jett BD, Huycke MM, Gilmore MS. Virulence of enterococci. Clin Microbiol Rev. 1994;7:462–78.PubMedGoogle Scholar
- Rice EW, Messer JW, Johnson CH, Reasoner DJ. Occurrence of high-level aminoglycoside resistance in environmental isolates of enterococci. Appl Environ Microbiol. 1995;61:374–6.PubMedGoogle Scholar
- Devriese LA, Pot B, Collins MD. Phenotypic identification of the genus Enterococcus and differentiation of phylogenetically distinct enterococcal species and species groups. J Appl Bacteriol. 1993;75:399–408.PubMedGoogle Scholar
- Willett HP. Energy metabolism. In: Joklik WK, Willett HP, Amos DB, Wilfert CM, editors. Zinsser microbiology. 20th ed. East Norwalk (CT): Appleton & Lange; 1992. p. 53-75.
- Ritchey TW, Seeley HW. Cytochromes in Streptococcus faecalis var. zymogenes grown in a haematin-containing medium. J Gen Microbiol. 1974;85:220–8.PubMedGoogle Scholar
- Pritchard GG, Wimpenny JWT. Cytochrome formation, oxygen-induced proton extrusion and respiratory activity in Streptococcus faecalis var. zymogenes grown in the presence of haematin. J Gen Microbiol. 1978;104:15–22.PubMedGoogle Scholar
- Ritchey TW, Seeley HW Jr. Distribution of cytochrome-like respiration in streptococci. J Gen Microbiol. 1976;93:195–203.PubMedGoogle Scholar
- Bryan-Jones DG, Whittenbury R. Haematin-dependent oxidative phosphorylation in Streptococcus faecalis. J Gen Microbiol. 1969;58:247–60.PubMedGoogle Scholar
- Williamson R. Le Bougu,nec C, Gutmann L, Horaud T. One or two low affinity penicillin-binding proteins may be responsible for the range of susceptibility of Enterococcus faecium to benzylpenicillin. J Gen Microbiol. 1985;131:1933–40.PubMedGoogle Scholar
- Bush LM, Calmon J, Cherney CL, Wendeler M, Pitsakis P, Poupard J, High-level penicillin resistance among isolates of enterococci: implications for treatment of enterococcal infections. Ann Intern Med. 1989;110:515–20.PubMedGoogle Scholar
- Sapico FL, Canawati HN, Ginunas VJ, Gilmore DS, Montgomerie JZ, Tuddenham WJ, Enterococci highly resistant to penicillin and ampicillin: an emerging clinical problem? J Clin Microbiol. 1989;27:2091–5.PubMedGoogle Scholar
- Horodniceanu T, Bougueleret L, El-Solh N, Bieth G, Delbos F. High-level, plasmid-borne resistance to gentamicin in Streptococcus faecalis subsp zymogenes. Antimicrob Agents Chemother. 1979;16:686–9.PubMedGoogle Scholar
- Zervos MJ, Kauffman CA, Therasse PM, Bergman AG, Mikesell TS, Schaberg DR. Nosocomial infection by gentamicin-resistant Streptococcus faecalis: an epidemiologic study. Ann Intern Med. 1987;106:687–91.PubMedGoogle Scholar
- Murray BE, Singh KV, Markowitz SM, Lopardo HA, Patterson JE, Zervos MJ, Evidence for clonal spread of a single strain of ß-lactamase-producing Enterococcus (Streptococcus) faecalis to six hospitals in five states. J Infect Dis. 1991;163:780–5.PubMedGoogle Scholar
- Uttley AHC, Collins CH, Naidoo J, George RC. Vancomycin-resistant enterococci. Lancet. 1988;1:57–8. DOIPubMedGoogle Scholar
- Leclercq R, Derlot E, Duval J, Courvalin P. Plasmid-mediated resistance to vancomycin and teicoplanin in Enterococcus faecium. N Engl J Med. 1988;319:157–61.PubMedGoogle Scholar
- Sahm DF, Kissinger J, Gilmore MS, Murray PR, Mulder R, Solliday J, In vitro susceptibility studies of vancomycin-resistant Enterococcus faecalis. Antimicrob Agents Chemother. 1989;33:1588–91.PubMedGoogle Scholar
- Arthur M, Courvalin P. Genetics and mechanisms of glycopeptide resistance in enterococci. Antimicrob Agents Chemother. 1993;37:1563–71.PubMedGoogle Scholar
- Clark NC, Cooksey RC, Hill BC, Swenson JM, Tenover FC. Characterization of glycopeptide-resistant enterococci from U.S. hospitals. Antimicrob Agents Chemother. 1993;37:2311–7.PubMedGoogle Scholar
- Klare I, Heier H, Claus H, Reissbrodt R, Van Witte W. A-mediated high-level glycopeptide resistance in Enterococcus faecium from animal husbandry. FEMS Microbiol Lett. 1995;125:165–72. DOIPubMedGoogle Scholar
- Noble WC, Virani Z, Cree RGA. Co-transfer of vancomycin and other resistance genes from Enterococcus faecalis NCTC 12201 to Staphylococcus aureus. FEMS Microbiol Lett. 1992;93:195–8. DOIGoogle Scholar
- Hiramatsu K, Hanaki H, Ino T, Yabuta K, Oguri T, Tenover FC. Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J Antimicrob Chemother. 1997;40:135–46. DOIPubMedGoogle Scholar
- Haley RW, Culver DH, White JW, Meade WM, Emori TG, Munn VP, The efficacy of infection surveillance and control programs in preventing nosocomial infections in US hospitals. Am J Epidemiol. 1985;121:182–205.PubMedGoogle Scholar
- Harris SL. Definitions and demographic characteristics. In: Kaye D, editor. Infective endocarditis. New York: Raven Press, Ltd.; 1992. p. 1-18.
- Hughes JM, Culver DH. W, Morgan WM, Munn VP, Mosser JL, Emori TG. Nosocomial infection surveillance, 1980-1982. MMWR Morb Mortal Wkly Rep 1983;32:1SS-16SS.
- Edmond MB, Ober JF, Dawson JD, Weinbaum DL, Wenzel RP. Vancomycin-resistant enterococcal bacteremia: natural history and attributable mortality. Clin Infect Dis. 1996;23:1234–9.PubMedGoogle Scholar
- Rhinehart E, Smith NE, Wennersten C, Gorss E, Freeman J, Eliopoulos GM, Rapid dissemination of ß-lactamase-producing, aminoglycoside-resistant Enterococcus faecalis among patients and staff on an infant-toddler surgical ward. N Engl J Med. 1990;26:1814–8.
- Chow JW, Kuritza A, Shlaes DM, Green M, Sahm DF, Zervos MJ. Clonal spread of vancomycin-resistant Enterococcus faecium between patients in three hospitals in two states. J Clin Microbiol. 1993;31:1609–11.PubMedGoogle Scholar
- Montecalvo MA, Horowitz H, Gedris C, Carbonaro C, Tenover FC, Issah A, Outbreak of vancomycin-, ampicillin-, and aminoglycoside-resistant Enterococcus faecium bacteremia in an adult oncology unit. Antimicrob Agents Chemother. 1994;38:1363–7.PubMedGoogle Scholar
- Livornese LL, Dias S, Samel C, Romanowski B, Taylor S, May P, Hospital-acquired infection with vancomycin-resistant Enterococcus faecium transmitted by electronic thermometers. Ann Intern Med. 1992;117:112–6.PubMedGoogle Scholar
- Handwerger S, Raucher B, Altarac D, Monka J, Marchione S, Singh KV, Nosocomial outbreak due to Enterococcus faecium highly resistant to vancomycin, penicillin, and gentamicin. Clin Infect Dis. 1993;16:750–5.PubMedGoogle Scholar
- Centers for Disease Control and Prevention. Recommendations for preventing the spread of vancomycin resistance: recommendations of the Hospital Infection Control Practices Advisory Committee (HICPAC). MMWR Morb Mortal Wkly Rep. 1995;44(No. RR-12):1–13.PubMedGoogle Scholar
- Morris JG Jr, Shay DK, Hebden JN, McCarter RJ Jr, Perdue BE, Jarvis W, Enterococci resistant to multiple antimicrobial agents, including vancomycin. Ann Intern Med. 1995;123:250–9.PubMedGoogle Scholar
- Edmond MB, Ober JF, Weinbaum DL, Pfaller MA, Hwang T, Sanford MD, Vancomycin-resistant Enterococcus faecium bacteremia: risk factors for infection. Clin Infect Dis. 1995;20:1126–33.PubMedGoogle Scholar
- Goldmann D, Larson E. Hand-washing and nosocomial infections. N Engl J Med. 1992;327:120–2.PubMedGoogle Scholar
- Noskin GA, Stosor V, Cooper I, Peterson LR. Recovery of vancomycin-resistant enterococci on fingertips and environmental surfaces. Infect Control Hosp Epidemiol. 1995;16:577–81. DOIPubMedGoogle Scholar
- Vollaard EJ, Clasener HAL. Colonization resistance. Antimicrob Agents Chemother. 1994;38:409–14.PubMedGoogle Scholar
- Quale J, Landman D, Saurina G, Atwood E, DiTore V, Patel K. Manipulation of a hospital antimicrobial formulary to control an outbreak of vancomycin-resistant enterococci. Clin Infect Dis. 1996;23:1020–5.PubMedGoogle Scholar
- Caron F, Pestel M, Kitzis M-D, Lemeland JF, Humbert G, Gutmann L. Comparison of different ß-lactam-glycopeptide-gentamicin combinations for an experimental endocarditis caused by a highly ß-lactam-resistant and highly glycopeptide-resistant isolate of Enterococcus faecium. J Infect Dis. 1995;171:106–12.PubMedGoogle Scholar
- Norris AH, Reilly JP, Edelstein PH, Brennan PJ, Schuster MG. Chloramphenicol for the treatment of vancomycin-resistant enterococcal infections. Clin Infect Dis. 1995;20:1137–44.PubMedGoogle Scholar
- Cohen MA, Yoder SL, Huband MD, Roland GE, Courtney CL. In vitro and in vivo activities of clinafloxacin, CI-990 (PD 131112), and PD 138312 versus enterococci. Antimicrob Agents Chemother. 1995;39:2123–7.PubMedGoogle Scholar
- Aumercier M, Bouhallab S, Capmau M-L, LeGoffic F. RP 59500: A proposed mechanism for its bactericidal activity. J Antimicrob Chemother. 1992;30(Suppl A):9–14.PubMedGoogle Scholar
- Collins LA, Malanoski GJ, Eliopoulos GM, Wennersten CB, Ferraro MJ, Moellering RC Jr. In vitro activity of RP59500, an injectable streptogramin antibiotic, against vancomycin-resistant gram-positive organisms. Antimicrob Agents Chemother. 1993;37:598–601.PubMedGoogle Scholar
- Chow JW, Davidson A, Sanford E III, Zervos MJ. Superinfection with Enterococcus faecalis during quinupristin/dalfopristin therapy. Clin Infect Dis. 1997;24:91–2.PubMedGoogle Scholar
- Chow JW, Donahedian SM, Zervos MJ. Emergence of increased resistance to quinupristin/dalfopristin during therapy for Enterococcus faecium bacteremia. Clin Infect Dis. 1997;24:90–1.PubMedGoogle Scholar
- Eliopoulos GM, Wennersten CB, Cole G, Moellering RC. In vitro activities of two glycylcyclines against gram-positive bacteria. Antimicrob Agents Chemother. 1994;38:534–41.PubMedGoogle Scholar
- Jones RN, Johnson DM, Erwin ME. In vitro antimicrobial activities and spectra of U-100592 and U-100766, two novel fluorinated oxazolidinones. Antimicrob Agents Chemother. 1996;40:720–6.PubMedGoogle Scholar
- Hillyard DR. The molecular approach to microbial diagnosis. Am J Clin Pathol. 1994;101:S18–21.PubMedGoogle Scholar
- Strohl WR. Biotechnology of Antibiotics. 2nd ed: Drugs and the Pharmaceutical Sciences 82, 1997.
- Jett BD, Jensen HG, Nordquist RE, Gilmore MS. Contribution of the pAD1-encoded cytolysin to the severity of experimental Enterococcus faecalis endophthalmitis. Infect Immun. 1992;60:2445–52.PubMedGoogle Scholar
- Ike Y, Hashimoto H, Clewell DB. High incidence of hemolysin production by Enterococcus (Streptococcus) faecalis strains associated with human parenteral infections. J Clin Microbiol. 1987;25:1524–8.PubMedGoogle Scholar
- Huycke MM, Spiegel CA, Gilmore MS. Bacteremia caused by hemolytic, high-level gentamicin-resistant Enterococcus faecalis. Antimicrob Agents Chemother. 1991;35:1626–34.PubMedGoogle Scholar
- Jett BD, Jensen HG, Atkuri R, Gilmore MS. Evaluation of therapeutic measures for treating endophthalmitis cause by isogenic toxin producing and toxin non-producing Enterococcus faecalis strains. Invest Ophthalmol Vis Sci. 1995;36:9–15.PubMedGoogle Scholar
- Ike Y, Hashimoto H, Clewell DB. Hemolysin of Streptococcus faecalis subspecies zymogenes contributes to virulence in mice. Infect Immun. 1984;45:528–30.PubMedGoogle Scholar
- Chow JW, Thal LA, Perri MB, Vazquez JA, Donabedian SM, Clewell DB, Plasmid-associated hemolysin and aggregation substance production contributes to virulence in experimental enterococcal endocarditis. Antimicrob Agents Chemother. 1993;37:2474–7.PubMedGoogle Scholar
- Ike Y, Clewell DB. Genetic analysis of pAD1 pheromone response in Streptococcus faecalis using transposon Tn917 as an insertional mutagen. J Bacteriol. 1984;158:777–83.PubMedGoogle Scholar
- Ike Y, Clewell DB, Segarra RA, Gilmore MS. Genetic analysis of the pAD1 hemolysin/bacteriocin determinant in Enterococcus faecalis: Tn917 insertional mutagenesis and cloning. J Bacteriol. 1990;172:155–63.PubMedGoogle Scholar
- Huycke MM, Gilmore MS. Frequency of aggregation substance and cytolysin genes among enterococcal endocarditis isolates. Plasmid. 1995;34:152–6. DOIPubMedGoogle Scholar
- Todd EW. A comparative serological study of streptolysins derived from human and from animal infections, with notes on pneumococcal haemolysin, tetanolysin and staphylococcus toxin. J Pathol Bacteriol. 1934;39:299–321. DOIGoogle Scholar
- Booth MC, Bogie CP, Sahl H-G, Siezen RJ, Hatter KL, Gilmore MS. Structural analysis and proteolytic activation of Enterococcus faecalis cytolysin, a novel lantibiotic. Mol Microbiol. 1996;21:1175–84. DOIPubMedGoogle Scholar
- Gilmore MS, Segarra RA, Booth MC. An hlyB-type function is required for expression of the Enterococcus faecalis hemolysin/bacteriocin. Infect Immun. 1990;58:3914–23.PubMedGoogle Scholar
- Katz L, Chu DT, Reich K. Bacterial genomics and the search for novel antibiotics. In: Plattner JJ, editor. Annual Reports in Medicinal Chemistry. Vol. 32. New York: Academic Press, Inc., 1997. p. 121-30.
- Shankar V, Gilmore MS. Structure and expression of a novel surface protein of Enterococcus faecalis. In: Abstracts of the 97th General Meeting of the American Society for Microbioloty; 4-8 May 1997;Miami Beach, Florida. Washington: The Society; 1997.
- Hancock LE, Gilmore MS. The contribution of a cell wall associated carbohydrate to the in vivo survival of Enterococcus faecalis in a murine model of infection. In: Abstracts of the 97th General Meeting of the American Society for Microbioloty. 4-8 May 1997;Miami Beach, Florida. Washington: The Society; 1997.
- Arduino RC, Murray BE, Rakita RM. Roles of antibodies and complement in phagocytic killing of enterococci. Infect Immun. 1994;62:987–93.PubMedGoogle Scholar
- Arduino RC, Palaz-Jacques K, Murray BE, Rakita RM. Resistance of Enterococcus faecium to neutrophil-mediated phagocytosis. Infect Immun. 1994;62:5587–94.PubMedGoogle Scholar
- Huycke MM, Joyce W, Wack MF. Augmented production of extracellular superoxide production by blood isolates of Enterococcus faecalis. J Infect Dis. 1996;173:743–6.PubMedGoogle Scholar
- Swartz MN. Hospital-acquired infections: diseases with increasingly limited therapies. Proc Natl Acad Sci U S A. 1994;91:2420–7. DOIPubMedGoogle Scholar
Figures
Cite This ArticleTable of Contents – Volume 4, Number 2—June 1998
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Michael Gilmore, Department of Microbiology and Immunology, University of Oklahoma Health Sciences Center, PO Box 26901, Oklahoma City, OK 73190, USA; fax: 405-271-8128
Top