Volume 11, Number 10—October 2005
Dispatch
Detecting Biological Warfare Agents
Cite This Article
Citation for Media
Abstract
We developed a fiber-optic, microsphere-based, high-density array composed of 18 species-specific probe microsensors to identify biological warfare agents. We simultaneously identified multiple biological warfare agents in environmental samples by looking at specific probe responses after hybridization and response patterns of the multiplexed array.
Accurately detecting and identifying biological warfare agents (BWAs) is the focal point for countering bioterrorism. Current methods used to identify BWAs are primarily focused on a single target agent (1–10). In contrast, the fiber-optic, microsphere-based, multiplexed arrays described in this paper can rapidly screen and simultaneously identify multiple potential BWAs, enabling an efficient response to a bioterrorism attack. Such multiagent analysis is difficult because it is usually complicated by interferences between assays and the large number of BWA probes.
We developed a platform to simultaneously detect BWAs with an array composed of DNA probe-functionalized microspheres that are randomly distributed onto microwells generated on the end of an etched coherent optical fiber bundle (11,12) that contains 6,000–50,000 individual fibers. Methods are described more fully in the Appendix. A multiplexed array was fabricated by distributing up to 18 different microsphere sensors into the optical fiber array with species-specific 50-mer DNA probes corresponding to 6 BWAs of interest (Bacillus anthracis, Yersinia pestis, Francisella tularensis, Brucella melitensis, Clostridium botulinum, and vaccinia virus) and 1 BWA simulant, Bacillus thuringiensis kurstaki, with each organism represented by at least 2 probe sequences (Table A1). All probe sequences were designed with software by Illumina, Inc. (San Diego, CA, USA), and the specificity of the probe sequences was checked against GenBank by using BLAST to have minimal cross-hybridization potential. Employing multiple probes for each BWA reduces the potential for both false-negative and false-positive results in environmental samples. The inherent redundancy of each probe type on the array, because each microsphere type has many replicates, provides additional confidence in detection and enhances the signal-to-noise ratio. Because the assembly process for the arrays is random, each bead type is encoded with an identifying optical bar code (11,12) created by entrapping dyes in various combinations within microspheres, to differentiate microsphere probe types from one another.
To ensure sensitive detection of BWAs with high accuracy, the performance of this multiplexed, high-density array was assessed by examining the detection limits and specificity of probes with Cy3-labeled synthetic targets (the synthetic target is a 50-mer oligonucleotide complementary to the probe). We modified our previous protocol of microsensor preparation (12) by using an increased concentration of cyanuric chloride solution in the probe activation step to achieve a more complete and efficient coupling of DNA probes to microspheres. The improved microsensor preparation enabled a detection limit as low as 10 fmol/L (in 50 μL volume) within 30 min hybridization time (data not shown). While the probes were selected to avoid the formation of secondary structures, such structures were not completely eliminated and probably affected the hybridization efficiency, resulting in higher detection limits for some probes on the array. To evaluate probe specificity, arrays containing beads with only a single probe type were exposed sequentially to the synthetic complementary target as well as to all noncomplementary targets. Cross-reactions were observed between probes from the same organism such as BA1/BA2, BA5/BA6, BTK1/BTK2, YP1/YP2, and FT1/FT2 (probes are abbreviated according to the organism from which they were derived: BA, B. anthracis; YP, Y. pestis; FT, F. tularensis; BM, Brucella melitensis, CB; C. botulinum; VA, vaccinia virus; and BTK, Bacillus thuringiensis kurstaki; details in Table A2) because some of the sequences for each probe pair overlapped, as probes were designed to be specific to the same target gene of the BWA. While in some applications cross-reactions are undesirable, in the present context the cross-reactions between probes from the same BWA confirmed each BWA in the sample. In addition, some minor cross-reactions between probe types from different BWAs were observed because of sequence similarity in spite of the careful probe design for each BWA (Table A2). These cross-reactions could be advantageous, since they generated a unique response pattern across all the probes on the multiplexed array for each target BWA and provided additional information to identify BWAs. As a result, any BWA can be simultaneously identified with this multiplexed array by looking at the high signals resulting from the specific response of each individual species-specific probe as well as the response pattern across the array.
Figure 1
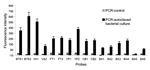
To sensitively detect target BWAs, polymerase chain reaction (PCR) was used as an amplification step before hybridization on the multiplexed array platform. Amplification was essential, especially when BWAs were present at trace levels in environmental samples (see below). Primer pairs that corresponded to each of the target sequences were designed specifically for each BWA (Table A1). Each primer pair amplified the respective BWA sequence from an autoclaved bacterial culture (Table A3) during asymmetric PCR. Figure 1 shows the detection of PCR products derived from each individual primer pair based on single bead–type arrays. The variation of the probe responses to PCR products can be explained by the different concentrations of the autoclaved bacterial cultures used as templates in PCR, the varying efficiency of primer pairs, or the potential of PCR products to form secondary structures. While carefully designed and optimized to have PCR products with a shorter length (147–201 bp), some of the amplified PCR fragments might form secondary structures (e.g., BA5). The formation of secondary structures makes the DNA fragments that were complementary to the array probe sequences inaccessible for hybridization. For these targets, the hybridization efficiency was affected, giving low hybridization responses to PCR products. In addition, the position of the target sequences within the PCR amplicons could affect hybridization efficiency. Oligonucleotide probes complementary to the middle part or the 5´ position of PCR products showed lower hybridization efficiencies than did target sequences close to the 3´ positions. This observation was likely related to steric hindrance that led to less accessibility of PCR products to the probe sequences on the array.
Figure 2

Figure 2. Gel analysis of multiplex polymerase chain reaction (PCR) with mixed autoclaved bacterial culture samples using primer pool I (A) and II (B). A) Lane 1, DNA ladder; lane 2, Bacillus anthracis...
Figure A1
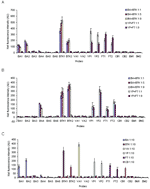
Figure A1. Simultaneous array detection of biological warfare agents in mixed autoclaved bacterial culture samples amplified with primer pool I (A), primer pool II (B), and wastewater samples spiked with primer pool I...
To assess the limit of detection with real bacterial samples, 10-fold serial dilutions were made from the autoclaved cultures of each BWA (Table A3) and used for PCR followed by array hybridization (summarized in the Table). Specificity tests of primer pairs were performed by applying each primer pair to other nontarget bacterial cultures individually, and no nonspecific amplifications were observed. To investigate the ability to simultaneously detect BWAs, multiplex PCR was used to analyze mixed bacterial cultures that contained multiple BWAs, e.g., B. anthracis mixed with B. thuringiensis kurstaki and Y. pestis mixed with F. tularensis, in varying ratios. We prepared 2 primer pools for the 7 BWAs of interest, and each pool contained 1 primer pair specific for each BWA. Figure 2 shows the amplification of BWAs in each mixed culture sample using primer pools I and II. The amplified PCR products were subjected to multiplexed array detection with each BWA represented by at least 2 probe types. The Figure A1 shows the results of simultaneous detection of BWAs in different mixed culture samples after amplification with primer pool I (Figure A1, panel A) and pool II (Figure A1, panel B). For the mixtures of B. anthracis and B. thuringiensis kurstaki, both probes BTK1 and BTK2 gave positive signals, as did probes BA1, BA2, and BA4, which indicates the presence of B. anthracis and B. thuringiensis kurstaki. Since the primer pair specific for B. anthracis in primer pool I and pool II corresponded to the BA2 and BA4 sequences, respectively, no positive signals were observed from the other BA probes (BA3, BA5, and BA6); no cross-reactions were seen between them except for probes BA1 and BA2 (Table A2). For mixtures of Y. pestis and F. tularensis, differentiating 2 bands (Figure 2B, lane 9 and lane 10) on the gel is difficult because the product size is similar; however, positive signals from the respective probes after hybridization showed these 2 BWAs in the mixed culture samples (Figure A1, panel B). Array detection in conjunction with multiplex PCR is advantageous in that it not only simultaneously identifies target BWAs but also minimizes the possibility of misinterpreting PCR results because it adds an additional level of specificity. With gel analysis only, PCR results are usually complicated because of nonspecific amplification during multiplex PCR with environmental samples.
Since real environmental samples will likely involve complex matrices with many potential interferents, the performance of the multiplexed array was further assessed with wastewater spiked with individual autoclaved bacterial cultures of BWAs at various volumetric dilution factors (1:3 and 1:10) (Table A3). Both primer pools were applied to all spiked wastewater samples for multiplex PCR. The probes on the multiplexed array gave positive responses only to the amplified PCR products of interest, without responding to any nonspecifically amplified products. Figure A1, panel C gives an example of array detection of the amplified BWAs from spiked wastewater samples (1:10 dilution) with primer pool I. BWAs seeded into wastewater at various dilution factors were all identified successfully within 30 minutes of hybridization.
In summary, this fiber-optic, microsphere-based array platform provides fast, sensitive, and simultaneous identification of BWAs with high accuracy. The high density of this array format can accommodate additional probe types while still maintaining a high redundancy for each probe type on the array. Additional probe types could be added to the array without affecting the performance of the existing microspheres. The ability to expand the probe types in the array opens up the opportunity to incorporate a large number of potential BWAs when their genome sequences become available. As a result, the multiplicity of arrays could be increased by incorporating an even broader class of potential BWAs and other pathogens, leading to a universal array for all pathogenic agents of interest.
Methods
Single Polymerase Chain Reaction (PCR)
A PCR Master Mix from Promega Corporation (Madison, WI, USA) was applied in all PCR assays. Asymmetric PCR was performed with a reaction mixture containing 2.5 U Taq DNA polymerase, a 200-μmol/L concentration of each deoxynucleoside triphosphate (dATP, dGTP, dCTP, and dTTP), 1.5 mmol/L MgCl2 in 1´ reaction buffer, 500 nmol/L Cy3 modified reverse primer, 50 nmol/L forward primer, and 10 μL DNA template from the autoclaved bacterial cultures. The amplification profile included an initial step at 95°C for 5 min, followed by 5 cycles of 95°C for 1 min, 56°C for 1 min, and 72°C for 1 min; 30 cycles of 95°C for 1 min, 54°C for 1 min, and 72°C for 1 min; and a final extension step at 72°C for 7 min. PCR controls were run with the reaction containing all the reagents for PCR amplification except template DNA.
Multiplex PCR
Primer pool I contained 7 primer pairs, BA2, BTK1, FT1, YP1, CB1, VA1, and BM1. Primer pool II contained BA4, BTK2, FT2, YP2, CB2, VA2, and BM2. Each primer pool has 1 primer pair for each biological warfare agent of interest. Multiplex PCR was run as follows: an initial step at 95°C for 5 min, followed by 5 cycles of 95°C for 1 min, 56°C for 1 min, and 72°C for 1 min; 40 cycles of 95°C for 1 min, 54°C for 1 min, and 72°C for 1 min; and a final extension step at 72°C for 7 min. PCR products were analyzed on 4% precast agarose gels (Invitrogen Corporation, Carlsbad, CA, USA) and visualized on Gel Doc XR System (Bio-Rad Laboratories, Hercules, CA, USA).
PCR products were denatured at 95°C for 5 min and chilled on ice. The denatured PCR products (25 μL) were combined with 10 μL hybridization buffer (2 mol/L NaCl and 0.2 mol/L phosphate-buffered saline [PBS]) and hybridized to the multiplexed array for 30 min at room temperature. Tris-EDTA (TE) buffer was used to wash the array after hybridization to remove the unbound target DNA, and 20% formamide in PBS (50°C) was applied for 1 min to minimize nonspecific binding of target DNA to probes followed by washing with TE buffer for another 1 min.
Ms Song is a graduate student of chemistry at Tufts University. Her research interests include applications of DNA microarrays and microarray-based detection of biological warfare agents and other infectious biological agents.
Acknowledgments
We are grateful to Illumina, Inc. for designing the probe sequences and primers and the Naval Medical Research Command for providing the autoclaved cultures of BWAs used in this study.
This work is funded by the Office of Naval Research through a grant from the Technical Support Working Group.
References
- Brightwell G, Pearce M, Leslie D. Development of internal controls for PCR detection of Bacillus anthracis. Mol Cell Probes. 1998;12:367–77. DOIPubMedGoogle Scholar
- Lee MA, Brightwell G, Leslie D, Bird H, Hamilton A. Fluorescent detection techniques for real-time multiplex strand-specific detection of Bacillus anthracis using rapid PCR. J Appl Microbiol. 1999;87:218–23. DOIPubMedGoogle Scholar
- Volokhov D, Rasooly A, Chumakov K, Chizhikov V. Identification of Listeria species by microarray-based assay. J Clin Microbiol. 2002;40:4720–8. DOIPubMedGoogle Scholar
- Volokhov D, Pomerantsev A, Kivovich V, Rasooly A, Chizhikov V. Identification of Bacillus anthracis by multiprobe microarray hybridization. Diagn Microbiol Infect Dis. 2004;49:163–71. DOIPubMedGoogle Scholar
- Belgrader P, Elkin CJ, Brown SB, Nasarabadi SN, Langlois RG, Milanovich FP, A reusable flow-through polymerase chain reaction instrument for the continuous monitoring of infectious biological agents. Anal Chem. 2003;75:3446–50. DOIPubMedGoogle Scholar
- Lindstrom M, Keto R, Markkula A, Nevas M, Hielm S, Korkeala H. Multiplex PCR assay for detection and identification of Clostridium botulinum types A, B, E, and F in food and fecal material. Appl Environ Microbiol. 2001;67:5694–9. DOIPubMedGoogle Scholar
- Yang JM, Bell J, Huang Y, Tirado M, Thomas D, Forster AH, An integrated, stacked microlaboratory for biological agent detection with DNA and immunoassays. Biosens Bioelectron. 2002;17:605–18. DOIPubMedGoogle Scholar
- Lagally ET, Scherer JR, Blazej RG, Toriello NM, Diep BA, Ramchandani M, Integrated portable genetic analysis microsystem for pathogen/infectious disease detection. Anal Chem. 2004;76:3162–70. DOIPubMedGoogle Scholar
- Ji J, Schanzle JA, Tabacco MB. Real-time detection of bacterial contamination in dynamic aqueous environments using optical sensors. Anal Chem. 2004;76:1411–8. DOIPubMedGoogle Scholar
- Tims TB, Lim DV. Rapid detection of Bacillus anthracis spores directly from powders with an evanescent wave fiber-optic biosensor. J Microbiol Methods. 2004;59:127–30. DOIPubMedGoogle Scholar
- Ferguson JA, Steemers FJ, Walt DR. High-density fiber-optic DNA random microsphere array. Anal Chem. 2000;72:5618–24. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 11, Number 10—October 2005
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
David R. Walt, 62 Talbot Ave, Medford, MA 02155, USA; fax: 617-627-3443
Top