Volume 13, Number 7—July 2007
Research
Ongoing Genome Reduction in Mycobacterium ulcerans
Cite This Article
Citation for Media
Abstract
Elucidation of the transmission, epidemiology, and evolution of Mycobacterium ulcerans, the causative agent of Buruli ulcer, is hampered by the striking lack of genetic diversity of this emerging pathogen. However, by using a prototype plasmid-based microarray that covered 10% of the genome, we found multiple genomic DNA deletions among 30 M. ulcerans clinical isolates of diverse geographic origins. Many of the changes appear to have been mediated by insertion sequence (IS) elements IS2404 and IS2606, which have high copy numbers. Classification of the deleted genes according to their biological functions supports the hypothesis that M. ulcerans has recently evolved from the generalist environmental M. marinum to become a niche-adapted specialist. The substantial genomic diversity, along with a prototype microarray that covered a small portion of the genome, suggests that a genome-wide microarray will make available a genetic fingerprinting method with the high resolution required for microepidemiologic studies.
The study of genetic diversity within bacterial species has provided information on aspects such as virulence (1,2), antimicrobial drug resistance (3), epidemiology, and microbial evolution (4–7). For mycobacteria such as Mycobacterium tuberculosis and M. ulcerans, low intraspecies diversity limits the use of genetic fingerprinting techniques that are based on sequence diversity in selected genetic elements. For M. tuberculosis, M. bovis, and the various bacillus Calmette-Guérin daughter strains, genome-wide microarray analyses have identified large sequence polymorphisms (4,8–10). However, the complete genome sequence of an organism is required for the design of synthetic oligonucleotide or PCR product–based microarrays. When this information is not available, an alternative is a PCR product–based shotgun DNA microarray (11), which we developed further into a plasmid-based microarray. We used this method for the differential genomic analysis of M. ulcerans, a human pathogen for which the fully assembled and annotated genome sequence was not available at the time of the study.
M. ulcerans is the causative agent of Buruli ulcer, an infectious disease characterized by chronic necrotizing skin ulcers (12). Buruli ulcer is an emerging infectious disease found mostly in West African countries but also in tropical and subtropical regions of Asia, the Western Pacific, and Latin America (13). Genetic analyses suggest recent divergence of M. ulcerans from M. marinum, a well-known fish pathogen that can cause limited granulomatous skin infections in humans (14). One of the hallmarks of the emergence of M. ulcerans as a more severe pathogen is the acquisition of a 174-kb plasmid that bears a cluster of genes necessary for the synthesis of the polyketide toxin mycolactone. This toxin appears largely responsible for the massive tissue destruction seen in Buruli ulcer (15). The epidemiology and mode of transmission of M. ulcerans disease are not fully understood, partly because no molecular typing method with sufficiently high resolution for microepidemiologic analyses is available.
Standard molecular typing methods such as multilocus sequence typing, restriction fragment length polymorphism, and fingerprinting using variable number of tandem repeats have shown an apparent lack of genetic diversity of M. ulcerans within individual geographic regions, which is indicative of a clonal population structure. The genotyping technique that has shown the highest discriminatory power so far is based on the use of outward-directed primers specific for the insertion sequence (IS) IS2404, in combination with an oligonucleotide that targets a repeated GC-rich motif (16). Application of this method determined the resolution of 10 different M. ulcerans genotypes, which correspond to the geographic origin of the isolates. However, this level of resolution is not sufficient for microepidemiologic analyses. We hypothesized that, as for M. tuberculosis (17), deletional and insertional events mediated by repetitive sequence elements are a major mechanism for genomic variation in M. ulcerans. To test this hypothesis, we developed a plasmid-based microarray and analyzed genomic DNA from 30 M. ulcerans isolates of diverse origins.
Plasmid-Based DNA Microarray
From a shotgun clone library of strain Agy99, 352 Escherichia coli plasmids (pCDNA2.1, Invitrogen, Basel, Switzerland) were randomly selected. Each plasmid contained an M. ulcerans DNA fragment of ≈2.3–2.7 kb. Given a genome size of 5,806 kb (18), this set of plasmid inserts represents a theoretical genome coverage of ≈10%. Plasmid DNA was prepared by using a Biomek 2000 Workstation (Beckman Coulter, Krefeld, Germany) and dissolved at a concentration of 150 ng/µL in 3× SSC (20× SSC stock solution is 3 M sodium chloride, 0.2 M sodium citrate, pH 7.0). The DNA samples were loaded on a piezo-dispensing head that contained 24 channels and spotted onto glass slides coated with poly-L-lysine (Superfrost Plus, Menzel, Braunschweig, Germany) by using a Topspot spotter (Biofluidix, Freiburg, Germany). Slides were incubated at 4ºC overnight and rehydrated under 50%–60% humidity for 1 h at room temperature. The spots resulting from a volume of ≈1 nL had an average diameter of 270 µm and were 500 µm apart from each other. The microarray layout displayed 2 identical fields—for hybridization with 2 different probes—that consisted of 2 replicates each, both of which contained 32 controls and 352 plasmids.
Biotinylation of M. ulcerans Genomic DNA Fragments
Figure 1

Figure 1. Distribution of outlier signals among the 30 Mycobacterium ulcerans isolates tested. M. ulcerans isolates: 1, 8756 Japan; 2, 980912 China; 3, 842 Suriname; 4, 7922 French Guiana; 5, 5147 Australia; 6,...
M. ulcerans clinical isolates used in this study are listed in Figure 1. Bacterial pellets of about 60 mg (wet weight) were heat inactivated for 1 h at 95°C in 500 µL extraction buffer (50 mmol/L Tris-HCl, 25 mmol/L EDTA, 5% monosodium glutamate) and sequentially treated with lysozyme (2 h, 37°C, 17 M lysozyme) and proteinase K (overnight, 45°C, 0.3 M proteinase K in proteinase K buffer: 1 mmol/L Tris-HCl, 5 mmol/L EDTA, 0.05% sodium dodecyl sulfate [SDS], pH 7.8). After digestion the samples were subjected to bead beater treatment (Mikro-Dismembrator, Braun Biotech International, Berlin, Germany) with 300 µL of 0.1-mm zirconia beads (BioSpec Products, Bartlesville, OK, USA) for 7 min at 3,000 rpm. DNA was extracted from the supernatants by phenol-chloroform (Fluka, Buchs, Switzerland) extraction and ethanol precipitation. Seven micrograms of M. ulcerans genomic DNA was digested with 3 U of Sau3A1 (New England Biolabs, Hitchin, UK) for 2 h at 37ºC and biotinylated according to Pollack et al. (19) using a BioPrime kit (Gibco/BRL, Gaithersburg, MD, USA). The biotinylated DNA was purified by using a Microcone YM-30 filter (Amicon/Millipore, Bedford, MA, USA), and its concentration was measured by optical density at 260 nm (GeneQuant spectrophotometer, Cambridge, UK).
Hybridization of Microarray Slides
Five micrograms of biotinylated DNA was mixed with 30 µg human Cot-1 DNA (Roche Applied Science, Indianapolis, IN, USA) and 100 µg yeast tRNA (Gibco/BRL). The hybridization mix was concentrated with a Speed Vac Concentrator System (Eppendorf, Basel, Switzerland), resolved in 3× SSC, 0.3% SDS, denatured for 3 min at 95ºC, and incubated for 30 min at 37ºC before hybridization. Microarray slides were cleaned with a nitrogen flow, exposed to UV light in a Stratalinker 2400 (Stratagene, La Jolla, CA, USA) at 650× 100 µJ, and heated for 5 min to 95ºC before application of 13 µL of the hybridization mix on each array field. Hybridization occurred for 20 h at 65ºC in a hydration chamber. Hybridized slides were washed once with 2× SSC, 0.03% SDS for 5 min at 65ºC, twice with 1× SSC for 5 min at room temperature, and finally with 0.2× SSC for 5 min at room temperature. The coloration step was performed with 2 mL staining solution containing 50% caseine, 1× maleic acid buffer (Roche Applied Science), and 2 µg Streptavidin Cy3 Fluorolink (Amersham, Piscataway, NJ, USA) for 30 min at room temperature, followed by additional washings for 5 min with 1× TBS (0.15 M sodium chloride, 0.02 M Tris, pH 7.5) as well as 0.1× TBS and drying with a nitrogen flow. DNA of all 30 M. ulcerans strains was processed under identical conditions and hybridized at least twice, which yielded 4 sets of data for each strain. Human Cot-1 DNA and plasmid DNA without insert as well as a hybridization mix without DNA served as negative controls for hybridization. A 500-bp β-lactamase gene fragment and Cy3-labeled random oligonucleotides (Microsynth, Balgach, Switzerland) were used as positive controls and for estimation of the amount of spotted DNA.
Microarray Scanning and Data Evaluation
Images of the microarrays were acquired by using a laser microarray scanner (GenePix 4100A, Axon Instruments Inc., Foster City, CA, USA) with an excitation wave length of 532 nm, an emission wavelength of 570 nm, and standardized measurement parameters. The resulting image was analyzed by the software GenePix Pro 4.1 (Axon Instruments Inc.), which enabled assignments of mean intensity values used for data interpretation. To select spots to be included in the analysis of genomic diversity of M. ulcerans strains, replicates of 10 hybridizations were performed by using M. ulcerans Agy99 genomic DNA. All spots that showed a signal lower than twice that given by the negative control plasmid without insert were rejected, as were all spots for which coefficient of variation was >30%. Further analysis used 232 spots that had an average signal above the threshold and sufficient signal stability. For each plasmid, we calculated the average signal value, standard deviation, and coefficient of variation and assessed a signal ratio in comparison with the reference strain. Outlier spots with a ratio higher than U2 (U2 = upper quartile + 3× interquartile) were identified through a box-plot analysis.
Characterization of Large Sequence Polymorphisms
Microarray data that indicated the presence of a deletion were verified by PCR analysis, which used primer pairs that spanned the insertion sequences of the respective plasmids, the flanking regions, or both. The 5′ and 3′ limits of the confirmed genomic deletions with respect to the genome of strain Agy99 were determined by PCR analysis, which used multiple sets of primers complementary to flanking genomic regions. PCR analyses that bridged the genomic breakpoints were performed by using a long-range PCR polymerase mix (Fermentas, St Leon-Rot, Germany) according to the manufacturer’s description. PCR products were cloned into pGEM-T (Catalys AG, Promega, Wallisellen, Switzerland) and sequenced using an ABI PRISM 310 genetic sequence analyzer (Perkin-Elmer, Waltham, MA, USA).
Comparative Genomic Hybridization of M. ulcerans Isolates
Figure 2
Figure 3

Figure 3. Box-plot analysis of the signal ratios obtained with genomic DNA of 30 Mycobacterium ulcerans strains identifying plasmids yielding outlier signals. Shown is the signal ratio in comparison with the African reference...
We constructed a microarray based on a random selection of 232 Escherichia coli plasmids obtained from a shotgun sequence library of the M. ulcerans isolate Agy99 from Ghana. Genomic DNA hybridization signal intensities from 30 M. ulcerans clinical isolates of worldwide distribution (Figure 2) were compared with those obtained with strain Agy99. Box-plot analysis (Figure 3) identified plasmids that yielded outlier signals with respect to strain Agy99. For 19 of 20 plasmids, PCR analysis confirmed an association of the outlier signal with a genomic deletion. Only 1 low hybridization signal represented a false-positive result (p188 from strain 940511, Côte d’Ivoire; Figure 3). The number of confirmed outlier plasmids per isolate ranged from zero for most African isolates to 9 for isolates from Suriname and French Guiana (Figure 1).
Figure 4
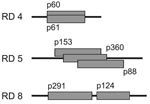
Figure 4. Positions of overlapping or adjacent plasmid inserts in regions of difference (RDs) 4, 5, and 8. Identical results retrieved by different plasmids with overlapping sequences or sequences in closest proximity demonstrate...
Of the 19 plasmid inserts that yielded confirmed outlier signals, 3 (p111, p299, and p341) contained sequences from the virulence plasmid pMUM001 of M. ulcerans. Of the 16 plasmids derived from the M. ulcerans chromosome, some contained fragments that overlapped the same region (Figure 4). Hybridizing regions were almost identical for p60 and p61. Both plasmids yielded outlier values with the isolates from Suriname and French Guiana. A cluster of overlapping inserts was observed for p88, p153, and p360; these produced outlier values for both of the Mexican isolates. The same pattern was seen with p124 and p291, which have inserts that are located in close proximity to each other in the genome (Figure 3). These results from related inserts demonstrated the reproducibility of the differential hybridization analysis. Because the inserts p60–p61, p88–p153–p360, and p124–p291 were part of the same deletion in regions of difference (RDs) 4, 5, and 8, respectively; (Figure 4), altogether 12 chromosomal RDs were identified.
Characterization of Genomic RDs
The 5′ and 3′ limits of the genomic deletions with respect to the genome of strain Agy99 were determined by PCR analysis that used multiple sets of primers complementary to plasmid inserts and to flanking genomic regions. The size of the deletions ranged from 1.8 kb to 53.1 kb (Table).
In 3 of the 12 RDs (RD3, 9, and 12), 2 distinct types of overlapping deletions (designated A and B) were observed, leading to a total of 15 large deletions. The overlapping deletions shared neither common 5′ nor 3′ end sequences. The strains from Australia had a 3.5-kb deletion in RD3; strains from Suriname and French Guiana had a slightly larger (3.8-kb) deletion. The isolates from Suriname and French Guiana had a larger (25.4-kb) deletion in RD9 than the isolates from Japan and China (17.7 kb). The largest deletion (53.1 kb) was designated RD12A and was observed in strains from Japan and China. Isolates from Suriname and French Guiana had a significantly smaller deletion in RD12 (35.2 kb). The 19.7-kb deletion 6 was found in isolates from 2 different regions (Mexico and Japan/China, respectively). All other deletions were observed in 2 isolates from the same region (Table).
To assess whether polymorphisms undetected by the microarray analysis would frequently occur in the identified RDs, we performed a detailed PCR analysis in all 30 M. ulcerans strains included in this study for 2 randomly selected RDs (RD5 and 12). We used 4 distinct primer pairs to span the insert sequence plus 5′ and 3′ flanking sequence stretches. For RD12, the PCR analysis confirmed the presence of a deletion in the 4 strains that had outlier signals in the microarray analysis, but no evidence for deletional polymorphism was obtained in the other strains. For RD5, PCR analysis confirmed the presence of a deletion in the 2 Mexican strains that had outlier signals (not shown). In addition, this PCR analysis identified the presence of an insertion in strains from Japan, China, Suriname, and French Guiana. The sequence of this 765-bp DNA insert was identical for all 4 strains. Its G+C content was 64%, and BLAST searches showed 98% identity with a sequence stretch of the M. marinum genome (www.sanger.ac.uk/cgi-bin/blast/submitblast/m_marinum) but no significant homology with sequences in the National Center for Biotechnology Information BLAST databases (www.ncbi.nlm.nih.gov/blast).
Association of Deletions with Insertions
Figure 5

Figure 5. Two distinct deletions in region of difference 3. Although deletion 3A represents a mere deletion event, the larger deletion 3B is associated with an insertion event. Neither 5’ nor 3’ ends...
Of the 15 identified genome rearrangement events, 1 (deletion 3A observed in 2 Australian isolates) was found to be a deletion, with the genomic sequences flanking the 5′ and 3′ borders of the 3,451-bp deletion being directly joined (Figure 5). Analysis of the other 14 deletions showed that the loss of DNA in a given strain with respect to the genome of Agy99 was associated with the insertion of substituting sequences of varying sizes unrelated to the deleted regions. As an example, the larger (3,784-bp) deletion 3B found in the isolates from Suriname and French Guiana was associated with the insertion of an unrelated DNA fragment, which comprised the 1,368 bp of IS2404 (20) plus an additional DNA stretch of 163 bp (Figure 5). For most of the other deletions, 1 of the 2 highly abundant insertion sequence elements (IS2404 or IS2606) was situated in either the genomic sequences that flanked the deletion or that were in the deleted parts or in the substituting sequence stretches (as for deletion 3B).
Analysis of Coding Sequences and Pseudogenes in the Deleted DNA Sequences
Figure 6

Figure 6. Functional categories of coding sequences (CDSs) and pseudogenes and their frequencies in the deletions and the whole genome. CDSs and pseudogenes in the identified regions of difference (RDs) and in the...
The 15 deletions identified contained 52 pseudogenes and 185 predicted protein-coding sequences (CDSs), which represent 5.7% of the annotated 4,143 CDSs in the genome of the M. ulcerans strain Agy99 (18). The number of deleted CDSs and pseudogenes ranged from 2 (RD4) to 50 (RD8 and 12A) and averaged 18.6 per deletion (Table). CDSs were classified into 11 functional categories (17). When compared with the gene composition of the entire Agy99 genome, the following functional categories were overrepresented among the 185 deleted CDSs: insertion sequences, unique hypothetical genes, and predicted proteins involved in detoxification (Figure 6). Also overrepresented was the deletion of the 52 pseudogenes that contain frame shift mutations and premature stop codons or that are disrupted by an insertion sequence. In contrast, genes involved in intermediary metabolism, information pathways, and cell wall/cell processes were underrepresented among the deleted CDSs (Figure 6). Of the 185 deleted functional CDSs, 89 had orthologs with >50% amino acid sequence identity to proteins from the M. tuberculosis H37Rv genome. A tendency for gene categories to cluster within the RDs was found. RD2 comprises 2 PPE genes: RDs 1, 12A, and 12B are predominantly CDSs involved in lipid metabolism, and RDs 9A and 11 include mainly transcriptional regulators. However, overall M. ulcerans lineages from distinct geographic origin (Africa, Australia, Asia, South America, Mexico) did not differ markedly in the categories of deleted genes. RD8 (deleted in the Mexican strains) is particularly interesting because it contains a cluster of proteins of the mammalian cell entry mce3 operon and associated regulators thereof. The transcriptional repressor, Mce3R, is considered to be an essential gene required for growth of M. tuberculosis (21). In addition, RD8 comprises a collection of CDSs of almost every functional category (Appendix Table). The spectrum of RD8-associated CDSs involved in detoxification included the multidrug transport protein mmr, the epoxide hydrolase EphB, the thiol peroxidase Tpx, and the alkyl hydroperoxide reductase C protein AhpC.
Although CDSs involved in intermediary metabolism were underrepresented among the deleted genes, 21 (42%) of deleted CDSs of this category were dehydrogenases (such as acyl-CoA short-chain alcohol, saccharopine, and aldehyde dehydrogenases), which are central enzymes in anaerobic metabolism (22) and important for survival in poorly oxygenated environments such as soil (23). In addition, other genes associated with anaerobic respiration, such as nitroreductases and electron transfer proteins, were found among the deleted CDSs.
We describe the use of a plasmid-based DNA microarray for identifying large deletional and insertional genomic polymorphisms in a collection of 30 M. ulcerans strains of geographically diverse origin. A set of plasmids randomly selected from an E. coli shotgun library of M. ulcerans genomic DNA was spotted on microarray slides. This is a newly developed technology, highly suitable for situations in which the complete genome sequence of a microorganism is not available. The prototype array used comprised 232 plasmids that yielded a reproducible and stable signal. Plasmids contained M. ulcerans genomic DNA fragments of 2.3–2.7 kb, thus reaching a theoretical genome coverage of 10%. Despite this incomplete coverage, 12 chromosomal and 3 virulence plasmid–associated RDs were identified. Fifteen distinct deletions of 1.8–53.1 kb were found and characterized in detail by sequence analysis within the 12 genomic RDs. The deletions identified were found in >1 M. ulcerans isolate, which demonstrates that they do not reflect events that occur during in vitro cultivation of individual isolates. The diversity of deletions within some genomic regions implies recombination hot spots or a selective advantage for loss of particular sequence stretches. Recombination events between adjacent copies of IS6110 in M. tuberculosis and IS100 in Yersinia pestis have been shown to promote the deletion of intervening DNA segments (9,23–26). Close association of RDs with the high copy number elements IS2404 and IS2606 of M. ulcerans indicates that these are involved in insertional and deletional events.
Although genome coverage with the prototype microarray used here was low, several geographic types of M. ulcerans could be differentiated. The largest group comprised all the African isolates (from Ghana, Benin, Côte d’Ivoire, Democratic Republic of Congo, Angola, and Togo), the isolates from Papua New Guinea, and some of the Australian isolates. A second group comprised the Australian strains 5142 and 5147, and a third group included the South American strains (from Suriname and French Guiana). The Mexican isolates represented a fourth; the Asian isolates (from Japan and China), a fifth subgroup. An extended analysis of insertions and deletions is expected to eventually give insight into the phylogenetic relationship between M. marinum and different lineages of M. ulcerans. Moreover, the use of a microarray that covers the whole genome may lead to the development of a genomic fingerprinting method, which is urgently needed for microepidemiologic studies that aim to characterize transmission pathways and environmental reservoirs of M. ulcerans.
The 15 distinct genomic deletions that we identified affected 6.2% of the M. ulcerans Agy99 genome, or 5.7% of the annotated CDSs and pseudogenes. When a whole-genome microarray was used to compare genomic DNA of 100 M. tuberculosis isolates, 5.5% of the genes were found to be affected (27). When one considers the limited genome coverage of the M. ulcerans prototype array used here, findings demonstrate a remarkably high degree of insertional and deletional diversity in M. ulcerans. In contrast, single nucleotide polymorphisms are rare (14).
Comparative genomic studies have shown that M. ulcerans recently evolved from the ubiquitous, fast-growing environmental bacterium M. marinum (www.sanger.ac.uk/projects/m_marinum) by lateral gene transfer and reductive evolution (18). Our comparative genomic hybridization analysis of a worldwide collection of M. ulcerans strains indicates that the downsizing of the genome from 6.6 Mb (M. marinum) to 5.8 Mb (M. ulcerans Agy99) is an ongoing process. Further genome reduction appears to be driving genetic diversification of M. ulcerans. Studies of other groups of microorganisms indicate that genome reduction is usually associated with adaptation to a more stable environment. An example is M. leprae, which has eliminated >2,000 genes upon adaptation to its human host (28). To which ecologic niche(s) in the environment or in host organisms M. ulcerans is adapting remains to be investigated.
Among the deleted CDSs are 11 members of the mammalian cell entry mce3 operon, which are regarded as virulence determinants in other mycobacteria. In M. tuberculosis the mce operons have been shown to code for genes important for entry and survival of the pathogen in mammalian cells (29,30). The 4 mce operons of M. tuberculosis have homologs among other mycobacteria. In particular, the mce3 operon has been found in M. avium and M. smegmatis; its deletion in M. bovis has been also documented (31). The 12.7-kb region that codes for the mce3 operon is located near the 3′ end of the RD2 element (32) that is present in M. bovis but absent in some strains of M. bovis BCG, which suggests the potential instability of this region. A mouse model of intradermal infection has recently shown that M. ulcerans is initially captured by phagocytes (33). In vitro studies suggest that the M. ulcerans intracellular stage is transient because phagocytic cells enter apoptosis-mediated cell death within 1 day. It will be interesting to investigate whether the mce3 operon plays a role during the transient invasion of host cells by M. ulcerans.
Overrepresentation of proteins involved in detoxification processes among the deleted CDSs indicates adaptation to a more stable environment. Deletion of many dehydrogenases thought to be involved in anaerobic respiration and of anaerobic respiratory enzymes and tranporters may give a hint that this niche is not anaerobic. At least in highly disease-endemic areas, M. ulcerans’ long-term persistence in chronic wounds and shedding into the environment may be relevant for the propagation of this species. Whether M. ulcerans is primarily adapting to persist in a specialized environmental habitat, in arthropod hosts (34), or in chronic wounds of mammalian hosts remains to be determined.
Dr Rondini is a microbiologist at the Swiss Tropical Institute, Basel, Switzerland. Her interests focus on the molecular microbiology of M. ulcerans.
Acknowledgments
We thank Adriana Ille for her technical assistance and Laura Gosoniu for her help in the statistical analysis, and we acknowledge the use of the BuruList web server (http://genopole.pasteur.fr/mulc/burulist.html) and M. marinum Blast server (www.sanger.ac.uk/cgi-bin/blast/submitblast/m_marinum).
This work was financed in part by the Stanley-Thomas-Johnson Foundation. M.K. was supported by a fellowship of the Deutsche Forschungsgemeinschaft (grant no. KA 1842/1-1).
References
- Hinchliffe SJ, Isherwood KE, Stabler RA, Prentice MB, Rakin A, Nichols RA, Application of DNA microarrays to study the evolutionary genomics of Yersinia pestis and Yersinia pseudotuberculosis.Genome Res. 2003;13:2018–29. DOIPubMedGoogle Scholar
- Perna NT, Plunkett GIII, Burland V, Mau B, Glasner JD, Rose DJ, Genome sequence of enterohaemorrhagic Escherichia coli O157:H7.Nature. 2001;409:529–33. DOIPubMedGoogle Scholar
- Kuroda M, Ohta T, Uchiyama I, Baba T, Yuzawa H, Kobayashi I, Whole genome sequencing of meticillin-resistant Staphylococcus aureus.Lancet. 2001;357:1225–40. DOIPubMedGoogle Scholar
- Behr MA, Wilson MA, Gill WP, Salamon H, Schoolnik GK, Rane S, Comparative genomics of BCG vaccines by whole-genome DNA microarray.Science. 1999;284:1520–3. DOIPubMedGoogle Scholar
- Colwell RR. Infectious disease and environment: cholera as a paradigm for waterborne disease.Int Microbiol. 2004;7:285–9.PubMedGoogle Scholar
- Hausdorff WP, Feikin DR, Klugman KP. Epidemiological differences among pneumococcal serotypes.Lancet Infect Dis. 2005;5:83–93.PubMedGoogle Scholar
- Perez-Perez GI, Rothenbacher D, Brenner H. Epidemiology of Helicobacter pylori infection.Helicobacter. 2004;9(Suppl1):1–6. DOIPubMedGoogle Scholar
- Cole ST. Comparative and functional genomics of the Mycobacterium tuberculosis complex.Microbiology. 2002;148:2919–28.PubMedGoogle Scholar
- Fleischmann RD, Alland D, Eisen JA, Carpenter L, White O, Peterson J, Whole-genome comparison of Mycobacterium tuberculosis clinical and laboratory strains.J Bacteriol. 2002;184:5479–90. DOIPubMedGoogle Scholar
- Gordon SV, Brosch R, Billault A, Garnier T, Eiglmeier K, Cole ST. Identification of variable regions in the genomes of tubercle bacilli using bacterial artificial chromosome arrays.Mol Microbiol. 1999;32:643–55. DOIPubMedGoogle Scholar
- Rathod PK, Ganesan K, Hayward RE, Bozdech Z, DeRisi JL. DNA microarrays for malaria.Trends Parasitol. 2002;18:39–45. DOIPubMedGoogle Scholar
- Asiedu K, Scherpbier R, Raviglione M. Buruli ulcer. Mycobacterium ulcerans infection. Geneva: World Health Organization, Global Buruli Ulcer Initiative; 2000.
- Johnson PD, Stinear T, Small PL, Pluschke G, Merritt RW, Portaels F, Buruli ulcer (M. ulcerans infection): new insights, new hope for disease control. PLoS Med. 2005;2:e108.Erratum in PLoS Med.2005;2:e173. DOIGoogle Scholar
- Stinear TP, Jenkin GA, Johnson PD, Davies JK. Comparative genetic analysis of Mycobacterium ulcerans and Mycobacterium marinum reveals evidence of recent divergence.J Bacteriol. 2000;182:6322–30. DOIPubMedGoogle Scholar
- Stinear TP, Mve-Obiang A, Small PL, Frigui W, Pryor MJ, Brosch R, Giant plasmid-encoded polyketide synthases produce the macrolide toxin of Mycobacterium ulcerans.Proc Natl Acad Sci U S A. 2004;101:1345–9. DOIPubMedGoogle Scholar
- Ablordey A, Kotlowski R, Swings J, Portaels F. PCR amplification with primers based on IS2404 and GC-rich repeated sequence reveals polymorphism in Mycobacterium ulcerans.J Clin Microbiol. 2005;43:448–51. DOIPubMedGoogle Scholar
- Brosch R, Pym AS, Gordon SV, Cole ST. The evolution of mycobacterial pathogenicity: clues from comparative genomics.Trends Microbiol. 2001;9:452–8. DOIPubMedGoogle Scholar
- Stinear TP, Seemann T, Pidot S, Frigui W, Reysset G, Garnier T, Reductive evolution and niche adaptation inferred from the genome of Mycobacterium ulcerans, the causative agent of Buruli ulcer.Genome Res. 2007;17:192–200. Epub 2007 Jan 8. DOIPubMedGoogle Scholar
- Pollack JR, Perou CM, Alizadeh AA, Eisen MB, Pergamenschikov A, Williams CF, Genome-wide analysis of DNA copy-number changes using cDNA microarrays.Nat Genet. 1999;23:41–6. DOIPubMedGoogle Scholar
- Stinear TP, Pryor MJ, Porter JL, Cole ST. Functional analysis and annotation of the virulence plasmid pMUM001 from Mycobacterium ulcerans.Microbiology. 2005;151:683–92. DOIPubMedGoogle Scholar
- Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis.Mol Microbiol. 2003;48:77–84. DOIPubMedGoogle Scholar
- Murugasu-Oei B, Tay A, Dick T. Upregulation of stress response genes and ABC transporters in anaerobic stationary-phase Mycobacterium smegmatis.Mol Gen Genet. 1999;262:677–82. DOIPubMedGoogle Scholar
- Kato-Maeda M, Rhee JT, Gingeras TR, Salamon H, Drenkow J, Smittipat N, Comparing genomes within the species Mycobacterium tuberculosis.Genome Res. 2001;11:547–54. DOIPubMedGoogle Scholar
- Brosch R, Gordon SV, Pym A, Eiglmeier K, Garnier T, Cole ST. Comparative genomics of the mycobacteria.Int J Med Microbiol. 2000;290:143–52.PubMedGoogle Scholar
- Deng W, Burland V, Plunkett GIII, Boutin A, Mayhew GF, Liss P, Genome sequence of Yersinia pestis KIM.J Bacteriol. 2002;184:4601–11. DOIPubMedGoogle Scholar
- Ho TB, Robertson BD, Taylor GM, Shaw RJ, Young DB. Comparison of Mycobacterium tuberculosis genomes reveals frequent deletions in a 20 kb variable region in clinical isolates.Yeast. 2000;17:272–82. DOIPubMedGoogle Scholar
- Tsolaki AG, Hirsh AE, DeRiemer K, Enciso JA, Wong MZ, Hannan M, Functional and evolutionary genomics of Mycobacterium tuberculosis: insights from genomic deletions in 100 strains.Proc Natl Acad Sci U S A. 2004;101:4865–70. DOIPubMedGoogle Scholar
- Cole ST, Eiglmeier K, Parkhill J, James KD, Thomson NR, Wheeler PR, Massive gene decay in the leprosy bacillus.Nature. 2001;409:1007–11. DOIPubMedGoogle Scholar
- Arruda S, Bomfim G, Knights R, Huima-Byron T, Riley LW. Cloning of an M. tuberculosis DNA fragment associated with entry and survival inside cells.Science. 1993;261:1454–7. DOIPubMedGoogle Scholar
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence.Nature. 1998;393:537–44. DOIPubMedGoogle Scholar
- Haile Y, Caugant DA, Bjune G, Wiker HG. Mycobacterium tuberculosis mammalian cell entry operon (mce) homologs in mycobacterium other than tuberculosis (MOTT).FEMS Immunol Med Microbiol. 2002;33:125–32. DOIPubMedGoogle Scholar
- Mahairas GG, Sabo PJ, Hickey MJ, Singh DC, Stover CK. Molecular analysis of genetic differences between Mycobacterium bovis BCG and virulent M. bovis.J Bacteriol. 1996;178:1274–82.PubMedGoogle Scholar
- Coutanceau E, Marsollier L, Brosch R, Perret E, Goossens P, Tanguy M, Modulation of the host immune response by a transient intracellular stage of Mycobacterium ulcerans: the contribution of endogenous mycolactone toxin.Cell Microbiol. 2005;7:1187–96. DOIPubMedGoogle Scholar
- Marsollier L, Robert R, Aubry J, Saint Andre JP, Kouakou H, Legras P, Aquatic insects as a vector for Mycobacterium ulcerans.Appl Environ Microbiol. 2002;68:4623–8. DOIPubMedGoogle Scholar
Figures
Table
Cite This ArticleTable of Contents – Volume 13, Number 7—July 2007
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Gerd Pluschke, Swiss Tropical Institute, Socinstr 57, CH 4002 Basel, Switzerland;
Top