Volume 14, Number 7—July 2008
Peer Reviewed Report Available Online Only
Toward a Unified Nomenclature System for Highly Pathogenic Avian Influenza Virus (H5N1)
Suggested citation for this article
Highly pathogenic avian influenza (HPAI) virus (H5N1) has appeared in >60 countries and continues to evolve and diversify at a concerning rate. Because different names have been used to describe emerging lineages of the virus, this study describes a unified nomenclature system to facilitate discussion and comparison of subtype H5N1 lineages.
The continuing geographic expansion and rapid evolution of HPAI subtype H5N1 virus across 3 continents is hindering control and eradication efforts in affected countries and raising public health concerns about a potential influenza pandemic. Since 1997, when the virus was discovered to cause disease and death in humans in Hong Kong, researchers have monitored the movement of the virus from region to region. Its molecular evolution has been characterized to better understand the spread of the virus and thus help prevent its perpetuation in poultry populations. Specific mutations and reassortment events that may enhance the virus’s ability to infect and be transmitted to humans (1–7) have also been scrutinized. Therefore, much effort has been spent to delineate the emerging lineages of the HPAI viruses (H5N1) from their earliest known progenitor, A/goose/Guangdong/96 (Gs/GD). From this ancestral virus, numerous lineages have evolved and because of rapid transcontinental spread, numerous publications have used different names to classify similar (if not identical) groups of viruses within the Gs/GD-like lineage (1–6). As a result, discussion and comparison of virus isolates have been hindered by a lack of uniformity in nomenclature, often leading to confusion in the interpretation of research results. The now routine practice of genome sequencing has also dramatically increased the sequence information available for analyses, adding to the complexity of examining the evolutionary relationships among HPAI virus (H5N1) isolates.
To address these issues, an international group of scientists and collaborators, referred to as the H5N1 Evolution Working Group, was convened at the Options for the Control of Influenza VI Conference in June 2007 in Toronto, Ontario, Canada. Their goal was to develop a unified nomenclature system for the classification of HPAI viruses (H5N1) belonging to the Gs/GD-like virus lineage. The initiative, which was encouraged and approved by 3 international agencies (the World Health Organization [WHO]), the World Organisation for Animal Health [OIE], and the Food and Agriculture Organization [FAO]), set out to unify the nomenclature system to simplify interpretation of sequence and surveillance data from different laboratories and to remove stigmatizing labeling of HPAI virus (H5N1) clades by geographic reference. Although most genes of the HPAI virus (H5N1) genome have undergone reassortment leading to their replacement by genes from lineages distinct from Gs/GD, the hemagglutinin (HA) protein gene has not been replaced since its emergence in 1996 (1). Therefore, monitoring the evolution of the Gs/GD HA lineage provides an initial constant by which H5N1 strains may be effectively compared. Taking these factors into account, we performed phylogenetic analyses on all of the publicly available subtype H5N1 HA sequences that have evolved in the Gs/GD lineage and designed a classification system. The results support the concept that the HPAI viruses (H5N1) currently circulating can be effectively grouped into multiple clades, herein designated by a hierarchical numbering system. Global adoption of the proposed H5 clade nomenclature and its expansion to other influenza lineages and genes, including other animal influenza virus subtypes, will benefit human and animal influenza research and public health.
Figure 1
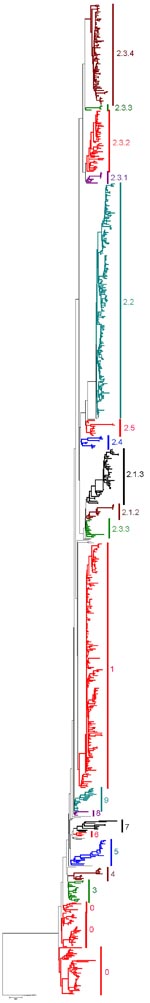
Figure 2
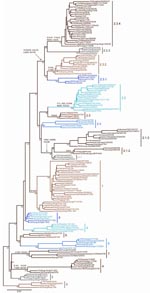
Nucleotide sequences of the HA gene of HPAI viruses (H5N1) were collected from publicly available databases: GenBank National Center for Biotechnology (NCBI) and the Influenza Sequence Database of Los Alamos National Laboratories (LANL). The analysis only included nearly full-length HA sequences (i.e., at least 1,600 nt in length) to ensure robust statistical support (Table 1). Multiple sequence alignment of 871 HA sequences was performed with ClustalW (www.ebi.ac.uk/Tools/clustalw2). The final alignment length was 1,707 nt. Isolates with 100% sequence similarity (i.e., redundant sequences) were identified and removed, giving a final alignment of 859 sequences. The appropriate DNA substitution model and γ-rate heterogeneity were determined with MrModeltest v2.2 (8) and used in all subsequent analyses. The neighbor-joining (NJ), maximum-likelihood (ML), and Bayesian methods used to construct trees for comparison are detailed in the legend of Figure 1. For ease of display, and also to ensure that the clade topology would be maintained if fewer isolates were used, a smaller dataset of 158 subtype H5N1 HA sequences was analyzed that included representative vaccine strains, reference serum strains, many human isolates, pathogenesis study strains, and geographically diverse isolates (Figure 2). Phylogenetic analyses were conducted on this dataset as described for Figure 1.
To quantify the nucleotide distances between and within groups identified on the phylogenetic tree, the average pairwise distances (between and within clades) were calculated by using MEGA version 3.1 (www.flu.org.cn/en/download-51.html) (13) by the NJ method with the Kimura 2-parameter model. Each distinct clade was determined to have an average distance >1.5% from other clades and an average distance <1.5% within the clade. Certain clades that comprise highly evolved HA genes depart slightly from these criteria; higher average intraclade distances were observed (i.e., Ck/Shanxi/2/2006 in clade 7).
Clade assignments were made by following several criteria used collectively to rationally name groups by a clade number. The criteria used to define clades are described in Table 1. Using these specific criteria, we identified and numbered 10 unique clades from the consensus topology of the large trees generated (Figure 1). The clade designations were then confirmed by the consensus topology of the smaller trees generated (Figures 1, 2). The topology of each clade was almost identical, and major clades were identified with consistency by using any of the 3 phylogenetic tree reconstruction methods (NJ, ML, and Bayesian). Also, the identified clades were consistent between the large and small datasets. Although the overall topologic organization between the large and small tree varied slightly (i.e., the positions of clades 7 and 8 are changed), the monophyletic grouping and bootstrap support for each clade remained predominately unchanged. However, trees derived from small datasets often yielded minor discrepancies; e.g., 4 isolates that were designated as clade 9 in the large tree were grouped with clade 8 in the smaller tree. The discrepancies in grouping between the large and small trees indicate the importance of using the largest datasets possible when classifying viral sublineages by phylogenetic analyses. Several of the identified clades were found to have distinct amino acid residues shared by members of that clade. To identify clade-specific amino acid residues, amino acid alignments were constructed for each clade, and residues shared by all members of that clade were compared with the Gs/GD/1/96 virus. Distinct shared amino acid residues are shown for each clade at the clade-defining node in Figure 2.
Using the clade designation criteria proposed in Table 1, this study has identified 10 unique first-order numbered clades of the HPAI viruses (H5N1) in the Gs/GD-like lineage (clades 0–9). The group of HA genes previously designated as clade 2 showed a level of diversity that far exceeds the current definition of a clade; therefore, this group was also separated into 5 additional second-order clades (clades 2.1–2.5). Clades 2.1 (avian/human isolates from Indonesia) and 2.3 (avian/human isolates from the People’s Republic of China; Hong Kong; Vietnam; Thailand; Lao People’s Democratic Republic; and Malaysia) were also further delineated into third-order groups (clades 2.1.1–2.1.3 and 2.3.1–2.3.4), respectively. The origins of isolates belonging to each clade are described in Table 2. For each clade identified, a representative prototype virus is listed to facilitate interpretation of the proposed numbering system (Table 2). As other studies have shown, the primary clade defining factor appears to be spatio-temporal because most distinct clades consist of isolates within close geographic proximity to one another or from specific time periods (perhaps as a result of heightened transmission during outbreak periods) (2–7). Notably, clade 2.2 comprises isolates from more widespread geographic areas (3 continents), which is likely to reflect movement of the virus through long-distance spread as a result of poultry trade or wild bird migration (2,3,6,7).
The evolution of the H5 HA in avian hosts shows a notable difference from the typical evolution of HA genes from human influenza viruses. The evolution of the H3 HA since 1968 is characterized by a limited diversity among circulating strains. This lack of diversity is clearly the consequence of rapid extinction after the emergence of new clades and lineages. As expected, the evolutionary tree of human influenza HA genes has extended trunks and extremely short branches (14,15). In contrast, multiple avian influenza A HA clades continue to evolve and co-circulate in different regions and species; hence, the unprecedented need for a nomenclature system that has been unnecessary for human influenza genes.
The results from this study indicate that the HPAI H5N1 viruses can be grouped into several clades designated by a numbering system that can continue to be expanded as these viruses continue to evolve. By establishing this nomenclature system and guidelines for naming clades, this information can be used in the future as criteria for assigning new clades as new lineages of HPAI H5N1 variants emerge.
Acknowledgment
We gratefully acknowledge the WHO, OIE, and the FAO for encouragement and support for this project. We also thank the research institutes, Ministries of Health, and Ministries of Agriculture from all of the countries that have contributed viral isolates or sequence data used in this study and for making this information publicly available in GenBank and LANL. Large and small trees containing publicly available sequences will be posted on the WHO GIP website (www.who.int/csr/disease/influenza/en) and the joint OIE-FAO network (OFFLU) website (www.offlu.net) and maintained as up-to-date (“evergreen”) evolutionary trees of the H5 HA to keep an open forum for following subtype H5N1 evolution.
References
- Duan L, Campitelli L, Fan XH, Leung YH, Vijaykrishna D, Zhang JX, Characterization of low-pathogenic H5 subtype influenza viruses from Eurasia: implications for the origin of highly pathogenic H5N1 viruses. J Virol. 2007;81:7529–39. DOIPubMedGoogle Scholar
- Ducatez MF, Olinger CM, Owoade AA, Tarnagda Z, Tahita MC, Sow A, Molecular and antigenic evolution and geographical spread of H5N1 highly pathogenic avian influenza viruses in western Africa. J Gen Virol. 2007;88:2297–306. DOIPubMedGoogle Scholar
- Chen H, Smith GJD, Li KS, Wang J, Fan XH, Rayner JM, Establishment of multiple sublineages of H5N1 influenza virus in Asia: Implications for pandemic control. Proc Natl Acad Sci U S A. 2006;103:2845–50. DOIPubMedGoogle Scholar
- Salzberg SL, Kingsford C, Cattoli G, Spiro DJ, Janies DA, Aly MM, Genome analysis linking recent European and African influenza (H5N1) viruses. Emerg Infect Dis. 2007;13:713–8.PubMedGoogle Scholar
- Smith GJD, Fan XH, Wang J, Li KS, Qin K, Zhang JX, Emergence and predominance of an H5N1 influenza variant in China. Proc Natl Acad Sci U S A. 2006;103:16936–41. DOIPubMedGoogle Scholar
- Smith GJD, Naipospos TSP, Nguyen TD, de Jong MD, Vijaykrishna D, Usman TB, Evolution and adaptation of H5N1 influenza virus in avian and human hosts in Indonesia and Vietnam. Virology. 2006;350:258–68. DOIPubMedGoogle Scholar
- Wallace RG, Hodac H, Lathrop RH, Fitch WM. A statistical phylogeography of influenza A H5N1. Proc Natl Acad Sci U S A. 2007;104:4473–8. DOIPubMedGoogle Scholar
- Nylander JAA. MRMODELTEST 2. Evolutionary Biology Centre, Uppsala University, Uppsala, Sweden; 2004 [cited 2007 Mar 12]. Available from http://www.abc.se/~nylander
- Swofford DL. PAUP: phylogenetic analysis using parsimony, version 4. Sunderland (MA): Sinauer Academic Publishers; 2001.
- Wood GW, Banks J, McCauley JW, Alexander DJ. Deduced amino acid sequences of the haemagglutinin of H5N1 avian influenza virus isolates from an outbreak in turkeys in Norfolk, England. Arch Virol. 1994;134:185–94. DOIPubMedGoogle Scholar
- Zwickl D. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion. PhD thesis, University of Texas at Austin; 2006 [cited 2008 Jun 5]. Available from http://www.bio.utexas.edu/faculty/antisense/garli/Garli.html
- Huelsenbeck JP, Ronquist FR. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–5. DOIPubMedGoogle Scholar
- Kumar S, Tamura K, Nei M. MEGA3: an integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief Bioinform. 2004;5:150–63. DOIPubMedGoogle Scholar
- Fitch WM, Leiter JM, Li XQ, Palese P. Positive Darwinian evolution in human influenza A viruses. Proc Natl Acad Sci U S A. 1991;88:4270–4. DOIPubMedGoogle Scholar
- Wolf YI, Viboud C, Holmes EC, Koonin EV, Lipman DJ. Long intervals of stasis punctuated by bursts of positive selection in the seasonal evolution of influenza A virus. Biol Direct. 2006;1:34. DOIPubMedGoogle Scholar
Figures
Tables
Suggested citation for this article: World Health Organization/World Organisation for Animal Health/Food and Agriculture Organization H5N1 Evolution Working Group. Toward a unified nomenclature system for highly pathogenic avian influenza virus (H5N1) [conference summary]. Emerg Infect Dis [serial on the Internet]. 2008 Jul [date cited]. Available from http://www.cdc.gov/EID/content/14/7/e1.htm
1The working group was established by request of the World Health Organization’s Global Influenza Programme, Department of Epidemic and Pandemic Alert and Response (WHO, GIP, EPR), the World Organisation for Animal Health (OIE), and the Food and Agriculture Organization (FAO). It consisted of the following members: Ruben O. Donis, Centers for Disease Control and Prevention (CDC), Atlanta, Georgia, USA (co-chair); Gavin J.D. Smith, University of Hong Kong, Hong Kong Special Administrative Region, People’s Republic of China (co-chair); Michael L. Perdue, WHO, GIP, EPR, Geneva, Switzerland2 (coordinator); Ian H. Brown, Veterinary Laboratories Agency, Addlestone, UK; Hualan Chen, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences CAAS, Harbin, People’s Republic of China; Ron A.M. Fouchier, Erasmus University, Rotterdam, the Netherlands; Yoshihiro Kawaoka, University of Wisconsin-Madison, Madison, Wisconsin, USA, and Institute of Medical Science, University of Tokyo, Tokyo, Japan; John Mackenzie, Curtin University of Technology, Perth, Western Australia, Australia; and Yuelong Shu, China Centers for Disease Control, Bejing, People’s Republic of China. In addition, the following persons made substantial contributions: Ilaria Capua, Instituto Zooprofilattico Sperimentale delle Venezie, Padova, Italy; Nancy Cox, Todd Davis, Rebecca Garten, and Catherine Smith, CDC; Yi Guan and Dhanasekaran Vijaykrishna, University of Hong Kong; Elizabeth Mumford, WHO, GIP, EPR; and Colin A. Russell and Derek Smith, University of Cambridge, Cambridge, UK.
2Current affiliation: US Department of Health and Human Services, Washington, DC, USA.
Table of Contents – Volume 14, Number 7—July 2008
| EID Search Options |
|---|
|
|
|
|
|
|