Volume 18, Number 4—April 2012
Research
Identification of Intermediate in Evolutionary Model of Enterohemorrhagic Escherichia coli O157
Cite This Article
Citation for Media
Abstract
Highly pathogenic enterohemorrhagic Escherichia coli (EHEC) O157 cause a spectrum of clinical signs that include diarrhea, bloody diarrhea, and hemolytic uremic syndrome. The current evolutionary model of EHEC O157:H7/H– consists of a stepwise evolution scenario proceeding from O55:H7 to a node (hypothetical intermediate) that then branches into sorbitol-fermenting (SF) O157:H– and non-SF (NSF) O157:H7. To identify this hypothetical intermediate, we performed single nucleotide polymorphism analysis by sequencing of 92 randomly distributed backbone genomic regions of 40 O157:H7/H– isolates. Overall, 111 single nucleotide polymorphisms were identified in 75/92 partial open reading frames after sequencing 51,041 nt/strain. The EHEC O157:H7 strain LSU-61 from deer occupied an intermediate position between O55:H7 and both O157 branches (SF and NSF O157), complementing the stepwise evolutionary model of EHEC O157:H7/H–. The animal origin of this intermediate emphasizes the value of nonhuman reservoirs in the clarification of the evolution of human pathogens.
Enterohemorrhagic Escherichia coli (EHEC) belongs to the Shiga toxin–producing E. coli group and causes clinical signs ranging from watery to bloody diarrhea for most symptomatically infected patients (1,2). EHEC serotypes O157:H7 and O157:H– (nonmotile) are the most frequently isolated from patients with severe EHEC-associated diseases, such as bloody diarrhea and hemolytic uremic syndrome. Infections caused by EHEC O157:H7/H– are major public health threats and require considerable resources for control and prevention (1,3). Sorbitol-fermenting (SF) EHEC O157:H–, initially found in Germany and later in other countries such as Scotland, Finland, and Australia, are increasingly associated with severe disease (4). These strains can ferment sorbitol after overnight incubation on sorbitol MacConkey agar, unlike non-SF (NSF) EHEC O157:H7. Today, SF EHEC O157:H– strains cause ≈20% of all hemolytic uremic syndrome cases in Germany (4–8). Classic NSF EHEC O157:H7 are of animal origin and have caused multiple outbreaks through contaminated food (4), but SF EHEC O157:H– are almost exclusively isolated from humans, which suggests that humans are the main reservoir (5).
On the basis of multilocus enzyme electrophoresis and multilocus sequence typing (MLST) data (9,10), the evolutionary model of EHEC O157 suggests that EHEC O157 emerged from E. coli O55:H7 by loss and acquisition of virulence and phenotypic traits (10). To further explain the evolution from O55:H7, a hypothetical intermediate and putatively extinct clone (missing link) SF O157:H7 emerging from O55:H7 was introduced; theoretically, it is from this intermediate that the 2 branches (NSF O157:H7 and SF O157:H–) diverged (9,10).
Shaikh and Tarr subdivided NSF O157:H7 into 3 clusters (11); in their analysis, the SF O157:H– branch remained evolutionary conserved and clearly separated from NSF O157:H7, with additional data suggesting a hypothetical intermediate. Recent studies based on whole core genome single-nucleotide polymorphisms (SNPs) enabled precise reconstruction of this model (12). The E. coli O157:H– strain LSU-61, which was isolated from a deer (10,13), had been previously discussed by Feng et al. as a potential intermediate, but that hypothesis was rejected because the strain lacked a gene encoding Shiga toxin (stx) and had a distinct MLST sequence type (10). We used an SNP-based approach to examine isolates from different sources of EHEC O157:H7/H– to further elucidate the evolutionary model of emergence of this pathogen, paying particular attention to identifying the “missing link” hypothetical intermediate.
Bacterial Strains Analyzed
Of the 50 EHEC strains examined (Table), 48 were serotype O157:H7/H– and 2 were O55:H7. Core or complete genome sequences were available for 8 O157 and 2 O55:H7 strains; these sequences served as a framework of the evolutionary model of EHEC O157. The remaining 40 strains consisted of 13 O157:H7/H– strains that represented different clusters according to previous multilocus variable-number tandem-repeat analysis (19); 26 O157:H7/H– strains isolated during 1987–2010 that were randomly chosen from our strain collection; and strain LSU-61, which was considered to be an intermediate (10).
Identification of EHEC O157 Strains
All 39 EHEC O157 isolates from our laboratory were isolated from stool samples as described (20,21). Isolates were confirmed to be E. coli by the API 20 Etest (bioMérieux, Marcy l’Etoile, France) and serotyped by using antiserum against E. coli O antigens 1–181 and H antigens 1–56 (22). Subtyping of fliC genes in nonmotile isolates by using HhaI restriction fragment-length polymorphism of amplicons obtained with primers FSa1 and rFSa1 (23,24) confirmed the presence of fliCH7 in all isolates. All strains were frozen at −70°C until further use.
Isolation of DNA
A single colony from a fresh overnight culture on Columbia blood agar (Heipha, Eppelheim, Germany) was inoculated into a liquid culture of nutrient broth medium (Heipha) and incubated overnight at 37°C. The liquid culture was used to prepare DNA as described (25), except that phenol extraction was omitted and the corresponding supernatants were directly precipitated with isopropanol.
Cluster Classification of O157:H7 Strains
Previously determined SNP patterns T/G/T/A or G/T/C/C at Sakai genome positions 337,933 (ECs0320, putative receptor), 1,460,599 (ECs1414, curli production assembly/transport component), 2,370,797 (ECs2397, transport system permease protein), and 5,404,166 (ECs5279, fimH–locus) have been shown to be cluster specific (12). On this basis, we used Sanger sequencing to group strains into cluster 3 or cluster 1 of subgroup C. Because the prototype strain of cluster 2 shared the SNP pattern with cluster 3, strains of cluster 2 were differentiated by using the published cluster differentiation scheme based on the occupancy of stx integration sites and the stx genotype (11,15). SNP pattern T/G/T/C was declared as unknown.
MLST and Sequencing of EHEC O157 Core Genomic Loci
As a first classification, we used MLST to determine the sequence type (ST)for all prototype strains of each subgroup and cluster by sequencing internal fragments of 7 housekeeping genes (adk, fumC, gyrB, icd, mdh, purA, and recA) (26). Alleles, STs, and clonal complexes were assigned in accordance with the E. coli MLST website (http://mlst.ucc.ie/mlst/dbs/Ecoli).
In addition to 10 SNP localizations that were known to differentiate subgroups and clusters (12), we randomly selected 82 additional backbone genomic regions for more in-depth SNP analysis. Using the Primer3 algorithm (http://frodo.wi.mit.edu/primer3), we developed 93 primer pairs that generated PCR products of backbone genomic regions ranging from 600 to 700 bp (Technical Appendix Table 1); for open reading frame (ORF) ECs3076, 2 separate primer pairs were designed to cover 2 described SNP localizations (12). EHEC O157:H7 strain Sakai served as a reference (GenBank accession no. NC_002655).
PCR was performed in a 14-µL reaction mixture containing 7 µL REDTaq (Sigma Aldrich, St. Louis, MO, USA), ≈6 ng DNA, and 1.5 µL each forward and reverse primer, with a final concentration of 10 µmol/L. The cycling reaction conditions were initial denaturation (2 min at 94°C), 35 cycles of denaturation (45 s at 94°C), annealing (60 s at 60°C), and extension (90 s at 72°C), followed by a final extension (10 min at 72°C). PCR products were purified by using the exonuclease I (New England Biolabs GmbH, Frankfurt-Hoechst, Germany) and shrimp alkaline phosphatase (USB Amersham, Freiburg, Germany) according to methods modified from (27). In brief, 7 µL of the PCR product was incubated simultaneously with 1.5 U of each enzyme at 37°C for 45 min, followed by enzyme heat inactivation at 80°C.
For sequencing of both strands, 2 µL of the purified amplicons was mixed with 0.5 µL premix from the ABI Prism BigDye Terminator v3.1 Ready Reaction Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) plus 1.8 µL Tris-HCl-MgCl2 buffer (400 mmol/L Tris-HCl, 10 mmol/L MgCl2; pH 9) and 2 µL (10 µmol/L) from the sequencing primer (forward or reverse primer, in a total volume of 10 µL. The cycling reaction conditions were 25 cycles of denaturation (10 s at 96°C) and combined annealing and extension (4 min at 60°C). Finally, the sequencing reaction products were purified by using an alcohol precipitation method as recommended by the manufacturer and loaded onto a 3130xl Genetic Analyzer (Applied Biosystems) for capillary sequencing.
Genotypic Characterization of LSU-61
To further evaluate the genotype of LSU-61 and its potential role in the evolutionary model of EHEC O157, we investigated known stx-phage integration sites. We used the draft genome sequence of the O157 strain LSU-61 (GenBank accession no. AEUC00000000) (28). yehV, a known integration site of stx1, was screened in silico by using primer pair A/B from (29). For analysis of the wrbA locus, a site of integration of the stx2 bacteriophage, we used primer pair C/D from (29). The 2 other currently known potential integration sites of stx2, yecE and sbcB, were screened by using primer pairs EC10/EC11, yecD-fwd/yecN-rev, and sbcB1/sbcB2 (30).
Data Analysis
Sequence trace files were analyzed and stored by using SeqSphere software version 0.9 beta (Ridom GmbH, Münster, Germany); a minimum-spanning tree was constructed with the integrated minimum-spanning tree algorithm. Gene functions were categorized by using the Pathosystems Resource Integration Center database (www.patricbrc.org/portal/portal/patric/Home) and corresponding Kyoto Encyclopedia of Genes and Genomes (KEGG; www.genome.jp/kegg) assignments. Overall, genes were grouped into 3 functional categories: metabolism/housekeeping, putative metabolism/housekeeping, and hypothetical protein. If no KEGG phenotype assignment was found, a putative metabolism/housekeeping function was predicted on the basis of BLAST (http://blast.ncbi.mlm.nih.gov/Blast.cgi) results.
Of the 48 EHEC O157 strains studied, 10 were SF serotype O157:H–. Subgrouping and cluster designation of the NSF O157 strains resulted in 18 cluster 1 strains, 1 cluster 2 strain, and 17 cluster 3 strains. For 2 strains, LSU-61 and SNPO157_36, no characteristic SNP pattern was determined (Table). Further characterization by MLST of prototype strains that defined subgroups and clusters resulted in identical STs for all SF and NSF O157 (ST11) and in closely related STs of the O55:H7 strains (ST335).
The 50 strains of serotypes O157:H7/H– and O55:H7 were further characterized with respect to their SNP prevalence in the core genome. In total, 92 core genomic loci were analyzed, comprising 51,041 bp sequencing information (≈0.9% of the O157:H7 Sakai genome) (Table; Technical Appendix Table 2). Sequencing demonstrated 111 biallelic variants, an average of 1.2 variants per sequenced locus (Technical Appendix Table 3). Deletions or insertions were not detected.
Of the 111 SNPs, 53 (47.7%) were synonymous SNPs (sSNPs) and 58 (52.3%) were nonsynonymous SNPs (nsSNPs); 78 (70.3%) SNPs were transitions, and 33 (29.7%) were transversions. Concatenation of all loci resulted in an average of 1 SNP every 460 bp; sSNPs occurred every 963 bp, and nsSNPs every 880 bp. On the level of analyzed partial ORFs, 1 SNP was found in 45 partial ORFs, 2 SNPs in 25 partial ORFs, 3 SNPs in 4 partial ORFs, and 4 SNPs in 1 partial ORF.
To further elucidate the SNP distribution, we categorized the 92 loci into 3 functional groups. Most loci belonged to (putative) metabolism or housekeeping genes because these were chosen solely from backbone regions. If no KEGG assignment was possible, we estimated the function of the corresponding fragment on the basis of BLAST homologies. Defined annotation information regarding the function in metabolism or housekeeping was determined for 25 partial ORFs. A housekeeping/metabolism function was predicted for 58 loci. The remaining 9 loci were hypothetical proteins only (Technical Appendix).
Figure
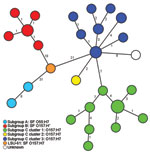
Figure. Minimum-spanning tree based on single-nucleotide polymorphism (SNP) genotypes illustrating the phylogeny of 50 enterohemorrhagic Escherichia coli O157:H7/H– and O55:H7 isolates and the intermediate position of strain LSU-61 during the evolution of...
On the basis of the 111 SNPs, the 50 strains were clustered into 27 SNP genotypes (Figure). The 111 SNPs were able to reconstruct the well-known evolutionary model (9–12) with the stepwise evolution from O55:H7 (subgroup A) to either SF O157:H– (subgroup B) or NSF O157:H7/H– (subgroup C, clusters 1–3). Strain LSU-61 interlinked all 3 subgroups, thereby substituting for the unknown intermediate (Figure). Moreover, the applied SNP scheme differentiated the O55:H7 strains and exhibited 5 genotypes within the 10 SF O157:H– strains segregated from the SF O157:H7 strain LSU-61. The NSF O157:H7 subgroup C1 strains comprising 18 isolates were differentiated in 9 SNP genotypes, and the subgroup C3 strains (n = 17) exhibited 8 SNP genotypes.
To further validate the role of stx-negative LSU-61 as a potential intermediate, we investigated each known potential stx insertion site in silico to determine the presence or absence of a Shiga toxin–carrying bacteriophage. We conducted BLAST searches within the recently published draft genome sequence of LSU-61 (28) by using published primers for the different insertion sites (29,30). All insertion sites for stx1 (yehV) and stx2 (wrbA, yecE, sbcB) were intact.
To investigate the effect of selective pressure on some loci and potential selecting biases, we analyzed sSNP and nsSNP types separately. In each scenario, the phylogenetic reconstruction resulted in comparable branching, with distinct lineages for SF and NSF O157 and strain LSU-61 as an intermediate. Only the number of SNP genotypes differed slightly: 19 sSNP genotypes (13 NSF O157:H7, 3 SF O157:H–, 2 O55:H7, and LSU-61) based on the 53 sSNPs and 22 nsSNP genotypes (16 NSF O157:H7, 3 SF O157:H–, 2 O55:H7, and LSU-61) based on the 58 nsSNPs. This excludes strong selection bias of the different loci.
On the basis of SNP analysis of 92 chromosomal backbone regions of EHEC O157, we identified an SF O157:H7 strain that complements the current model of the stepwise evolution from O55:H7 to EHEC O157 in which the hypothetical intermediate between O55:H7 and SF and NSF O157:H7/H– has been unknown (10,12). As with the highly human pathogenic O157:H7 lineage of EHEC, which is known to reside in cattle, deer, and other ruminants, this intermediate strain was isolated from a deer (13). These findings support previous observations (31,32) and suggest an evolution toward an animal reservoir for O157:H7 soon after O157:H– and O157:H7 divergence. Strain LSU-61 is motile (H-phase 7) and enterohemolysin active (10), traits that are typical for NSF O157, further suggesting the intermediate character of LSU-61 between SF and NSF O157. In contrast to the MLST scheme applied from Feng et al. and Lacher et al. (10,33), our MLST analysis, using the scheme of Wirth et al. (26) that analyzes different genes, further corroborates the intermediate character of LSU-61 because it shares the same ST with SF (subgroup B prototype strain) and NSF O157 (subgroup C prototype strains).
Strain LSU-61 does not carry a stx gene, but this fact does not contradict our findings because these genes are encoded on bacteriophages that can be acquired and lost (30,34,35), and we do not have evidence of a progenitor to LSU-61 that contains stx genes. Although known potential stx phage integration sites in O157 were intact, the possibility of a previous stx bacteriophage carriage cannot be excluded. If the SF O157:H7 cluster emerged ≈3,000–4,000 years ago (12), certain genetic and phenotypic changes (10) occurred well before the first descendants of this cluster were isolated and characterized.
Two previous studies (31,32) reported isolated comparable strains to LSU-61 from (European) red deer, belonging to the same family (Cervidae) as white-tailed deer (North America), with comparable phenotypic and genotypic traits. Some of these were SF O157:H7 strains (stx negative or positive, β-glucuronidase positive activity) (31,32). The proof of the existence of SF O157:H7 in a ruminant (deer) host may indicate transfer into animals soon after the 2 (human pathogenic) O157 subgroups B and C emerged. On the basis of shared characteristics with both O157 branches, we suggest strain LSU-61 as a representative of the intermediate cluster complementing the stepwise evolutionary model of EHEC O157. The phylogeny based on either sSNPs or nsSNPs also resulted in a comparable phylogenetic tree with LSU-61 as a member of the progenitor node, underlining its intermediate role.
On the level of gene categories, a higher percentage of sSNPs, though fewer SNPs overall, were observed in the metabolism/housekeeping category compared with the putative metabolism/housekeeping category. The higher rate of nsSNPs in the latter category, resulting in a higher phenotypic diversity, might be explained by uncertain gene categorization because of currently limited knowledge of gene function. Therefore, SNP typing results may help to find genes involved in host–pathogen interactions rather than in metabolism or housekeeping only. SNP data for hypothetical proteins are difficult to interpret because information about their function is too imprecise to enable estimation of the effect of evolutionary pressure.
The fact that 35 of the 38 O157:H7 strains were subgrouped into either cluster 1 or 3 (17 and 18 strains, respectively) shows a certain persistence of these O157:H7 clusters (29), characterized by a successful pathogenicity, for example, outbreaks over a broad time frame (4). The preponderance of cluster 1 strains has been noted before, as have the paucity of cluster 2 and the diminished proportion of cluster 3 strains in North America (29). We observed a higher number of SNPs within the different NSF O157:H7 clusters compared with the few SNPs within restricted SF O157:H– genotypes and a maximum pairwise distance of 2 SNPs (Figure). A reason for this phenomenon may be the different animal host origins for the NSF O157:H7 clade, whereas SF O157:H– are considered to have only 1 main host, humans (5,19). This high conservation was similarly recognized when multilocus variable-number tandem-repeat analysis was applied (19). In this context, certain SNP genotypes may serve to illuminate several strain-specific characteristics, such as increased virulence and other phenotypic traits, as other studies have similarly observed for both SF and NSF O157 (36,37).
Our results could be interpreted as if C2 strain 86–24 is an offshoot of cluster 3, which is in contrast to the established stepwise model of O157. However, we believe that this is an artifact caused by sampling bias of the investigated 92 loci because only 11 backbone SNPs have been found to differentiate cluster 2 and 3 within the whole chromosomal backbone (12). One strain (SNPO157_36) did not cluster into any known O157:H7 cluster (Figure).
In summary, our identification of an intermediate member of the EHEC 1 clade complements the current evolutionary model of EHEC O157 by using chromosomal backbone SNP data of a spatiotemporally diverse strain collection. The different levels of genotypic conservation within the subgroups and the animal origin of the intermediate underline the great effect of host–pathogen interaction on the evolution of bacterial species. Future studies should focus on this interaction within both human and animal hosts to understand the evolution and persistence in nature of such human pathogens. The survival of the ancestral pathogen until today suggests that its genetic attributes could be informative in identifying fitness and potentially pathogenic loci.
Dr Jenke is a scientist at the Institute for Hygiene at the University Hospital Münster. His research interests include molecular typing and the epidemiology and phylogeny of Shiga toxin–producing E. coli.
Acknowledgments
We thank Peter Feng for providing strain LSU-61 for this analysis and Phillip I. Tarr for critical reading of the manuscript.
This study was supported by grants from the German Federal Ministry of Education and Research (nos. 0315219A and 01KI0801), from the EU Network ERA-NET PathoGenoMics II (no. 0315443), and from the medical faculty of the University Münster (no. BD9817044).
References
- Holtz LR, Neill MA, Tarr PI. Acute bloody diarrhea: a medical emergency for patients of all ages. Gastroenterology. 2009;136:1887–98. DOIPubMedGoogle Scholar
- Levine MM. Escherichia coli that cause diarrhea: enterotoxigenic, enteropathogenic, enteroinvasive, enterohemorrhagic, and enteroadherent. J Infect Dis. 1987;155:377–89. DOIPubMedGoogle Scholar
- Tarr PI, Gordon CA, Chandler WL. Shiga toxin–producing Escherichia coli and haemolytic uraemic syndrome. Lancet. 2005;365:1073–86. DOIPubMedGoogle Scholar
- Karch H, Tarr PI, Bielaszewska M. Enterohaemorrhagic Escherichia coli in human medicine. Int J Med Microbiol. 2005;295:405–18. DOIPubMedGoogle Scholar
- Karch H, Bielaszewska M. Sorbitol-fermenting Shiga toxin–producing Escherichia coli O157:H– strains: epidemiology, phenotypic and molecular characteristics, and microbiological diagnosis. J Clin Microbiol. 2001;39:2043–9. DOIPubMedGoogle Scholar
- Karch H, Mellmann A, Bielaszewska M. Epidemiology and pathogenesis of enterohaemorrhagic Escherichia coli. Berl Munch Tierarztl Wochenschr. 2009;122:417–24.PubMedGoogle Scholar
- Werber D, Bielaszewska M, Frank C, Stark K, Karch H. Watch out for the even eviler cousin—sorbitol-fermenting E coli O157. Lancet. 2011;377:298–9. DOIPubMedGoogle Scholar
- Feng P, Lampel KA, Karch H, Whittam TS. Genotypic and phenotypic changes in the emergence of Escherichia coli O157:H7. J Infect Dis. 1998;177:1750–3. DOIPubMedGoogle Scholar
- Feng PCH, Monday SR, Lacher DW, Allison L, Siitonen A, Keys C, Genetic diversity among clonal lineages within Escherichia coli O157:H7 stepwise evolutionary model. Emerg Infect Dis. 2007;13:1701–6.PubMedGoogle Scholar
- Shaikh N, Tarr PI. Escherichia coli O157:H7 Shiga toxin–encoding bacteriophages: integrations, excisions, truncations, and evolutionary implications. J Bacteriol. 2003;185:3596–605. DOIPubMedGoogle Scholar
- Leopold SR, Magrini V, Holt NJ, Shaikh N, Mardis ER, Cagno J, A precise reconstruction of the emergence and constrained radiations of Escherichia coli O157 portrayed by backbone concatenomic analysis. Proc Natl Acad Sci U S A. 2009;106:8713–8.PubMedGoogle Scholar
- Dunn JR, Keen JE, Moreland D, Alex T. Prevalence of Escherichia coli O157:H7 in white-tailed deer from Louisiana. J Wildl Dis. 2004;40:361–5.PubMedGoogle Scholar
- Perna NT, Plunkett G III, Burland V, Mau B, Glasner JD, Rose DJ, Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature. 2001;409:529–33. DOIPubMedGoogle Scholar
- Shaikh N, Holt NJ, Johnson JR, Tarr PI. Fim operon variation in the emergence of enterohemorrhagic Escherichia coli: an evolutionary and functional analysis. FEMS Microbiol Lett. 2007;273:58–63. DOIPubMedGoogle Scholar
- Karch H, Wiss R, Gloning H, Emmrich P, Aleksic S, Bockemühl J. Hemolytic-uremic syndrome in infants due to verotoxin-producing Escherichia coli [in German]. Dtsch Med Wochenschr. 1990;115:489–95. DOIPubMedGoogle Scholar
- Kulasekara BR, Jacobs M, Zhou Y, Wu Z, Sims E, Saenphimmachak C, Analysis of the genome of the Escherichia coli O157:H7 2006 spinach-associated outbreak isolate indicates candidate genes that may enhance virulence. Infect Immun. 2009;77:3713–21. DOIPubMedGoogle Scholar
- Hayashi T, Makino K, Ohnishi M, Kurokawa K, Ishii K, Yokoyama K, Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 2001;8:11–22. DOIPubMedGoogle Scholar
- Jenke C, Harmsen D, Weniger T, Rothgänger J, Hyytiä-Trees E, Bielaszewska M, Phylogenetic analysis of enterohemorrhagic Escherichia coli O157, Germany, 1987–2008. Emerg Infect Dis. 2010;16:610–6.PubMedGoogle Scholar
- Friedrich AW, Bielaszewska M, Zhang W, Pulz M, Kuczius T, Ammon A, Escherichia coli harboring Shiga toxin 2 gene variants: frequency and association with clinical symptoms. J Infect Dis. 2002;185:74–84. DOIPubMedGoogle Scholar
- Mellmann A, Bielaszewska M, Zimmerhackl LB, Prager R, Harmsen D, Tschäpe H, Enterohemorrhagic Escherichia coli in human infection: in vivo evolution of a bacterial pathogen. Clin Infect Dis. 2005;41:785–92. DOIPubMedGoogle Scholar
- Prager R, Strutz U, Fruth A, Tschäpe H. Subtyping of pathogenic Escherichia coli strains using flagellar (H)-antigens: serotyping versus fliC polymorphisms. Int J Med Microbiol. 2003;292:477–86. DOIPubMedGoogle Scholar
- Sonntag AK, Prager R, Bielaszewska M, Zhang W, Fruth A, Tschäpe H, Phenotypic and genotypic analyses of enterohemorrhagic Escherichia coli O145 strains from patients in Germany. J Clin Microbiol. 2004;42:954–62. DOIPubMedGoogle Scholar
- Zhang Y, Laing C, Steele M, Ziebell K, Johnson R, Benson AK, Genome evolution in major Escherichia coli O157:H7 lineages. BMC Genomics. 2007;8:121. DOIPubMedGoogle Scholar
- Wilson K. Preparation of genomic DNA from bacteria. Curr Protoc Mol Biol. 2001;Chapter 2:Unit 2.4.
- Wirth T, Falush D, Lan R, Colles F, Mensa P, Wieler LH, Sex and virulence in Escherichia coli: an evolutionary perspective. Mol Microbiol. 2006;60:1136–51. DOIPubMedGoogle Scholar
- Dugan KA, Lawrence HS, Hares DR, Fisher CL, Budowle B. An improved method for post-PCR purification for mtDNA sequence analysis. J Forensic Sci. 2002;47:811–8.PubMedGoogle Scholar
- Rump LV, Strain EA, Cao G, Allard MW, Fischer M, Brown EW, Draft genome sequences of six Escherichia coli isolates from the stepwise model emergence of Escherichia coli O157:H7. J Bacteriol. 2011;193:2058–9. DOIPubMedGoogle Scholar
- Besser TE, Shaikh N, Holt NJ, Tarr PI, Konkel ME, Malik-Kale P, Greater diversity of Shiga toxin–encoding bacteriophage insertion sites among Escherichia coli O157:H7 isolates from cattle than in those from humans. Appl Environ Microbiol. 2007;73:671–9. DOIPubMedGoogle Scholar
- Bielaszewska M, Prager R, Zhang W, Friedrich AW, Mellmann A, Tschäpe H, Chromosomal dynamism in progeny of outbreak-related sorbitol-fermenting enterohemorrhagic Escherichia coli O157:NM. Appl Environ Microbiol. 2006;72:1900–9. DOIPubMedGoogle Scholar
- García-Sánchez A, Sanchez S, Rubio R, Pereira G, Alonso JM, Hermoso de Mendoza J, Presence of Shiga toxin–producing E. coli O157:H7 in a survey of wild artiodactyls. Vet Microbiol. 2007;121:373–7. DOIPubMedGoogle Scholar
- Díaz S, Vidal D, Herrera-Leon S, Sanchez S. Sorbitol-fermenting, β-glucuronidase–positive, Shiga toxin–negative Escherichia coli O157:H7 in free-ranging red deer in south-central Spain. Foodborne Pathog Dis. 2011;8:1313–5. DOIPubMedGoogle Scholar
- Lacher DW, Steinsland H, Blank TE, Donnenberg MS, Whittam TS. Molecular evolution of typical enteropathogenic Escherichia coli: clonal analysis by multilocus sequence typing and virulence gene allelic profiling. J Bacteriol. 2007;189:342–50. DOIPubMedGoogle Scholar
- Bielaszewska M, Köck R, Friedrich AW, von Eiff C, Zimmerhackl LB, Karch H, Shiga toxin–mediated hemolytic uremic syndrome: time to change the diagnostic paradigm? PLoS ONE. 2007;2:e1024. DOIPubMedGoogle Scholar
- Mellmann A, Lu S, Karch H, Xu J, Harmsen D, Schmidt MA, Recycling of Shiga toxin 2 genes in sorbitol-fermenting enterohemorrhagic Escherichia coli O157:NM. Appl Environ Microbiol. 2008;74:67–72. DOIPubMedGoogle Scholar
- Manning SD, Motiwala AS, Springman AC, Qi W, Lacher DW, Ouellette LM, Variation in virulence among clades of Escherichia coli O157:H7 associated with disease outbreaks. Proc Natl Acad Sci U S A. 2008;105:4868–73. DOIPubMedGoogle Scholar
- Zhang W, Qi W, Albert TJ, Motiwala AS, Alland D, Hyytiä-Trees EK, Probing genomic diversity and evolution of Escherichia coli O157 by single nucleotide polymorphisms. Genome Res. 2006;16:757–67. DOIPubMedGoogle Scholar
Figure
Table
Cite This ArticleTable of Contents – Volume 18, Number 4—April 2012
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Alexander Mellmann, Institut für Hygiene, Universitätsklinikum Münster, Robert-KocH–Strasse 41, 48149 Münster, Germany
Top