Volume 18, Number 6—June 2012
Dispatch
Genome Analysis of Rift Valley Fever Virus, Mayotte
Cite This Article
Citation for Media
Abstract
As further confirmation of a first human case of Rift Valley fever in 2007 in Comoros, we isolated Rift Valley fever virus in suspected human cases. These viruses are genetically closely linked to the 2006–2007 isolates from Kenya.
Identified during the 1930s in Kenya, Rift Valley fever (RVF) is a zoonotic disease that circulates in many African countries and in the Arabian Peninsula (1,2). RVF virus (RVFV) epizootics are characterized by large sweeping abortion storms and substantial death rates in adult livestock (primarily sheep, goats, and cattle), with the death rate for newborn animals approaching 100% (3). Humans typically are infected by bites of infected mosquitoes or by percutaneous or aerosol exposure to contaminated fluids from infected animals. In most human cases, RVF is characterized by a self-limiting febrile illness (2–5 days), which progresses to more serious complications (hepatitis, encephalitis, blindness, or hemorrhagic syndrome) in only 1%–2% of infected persons (4,5). A large epizootic–epidemic occurred during 2006–2007 on the eastern African mainland, predominantly in Kenya (6) and Madagascar, during 2 successive rainy seasons (7).
In July 2007, a 12-year-old boy with a 2-month history of severe encephalitis was transferred from the Grande Comore, Union of the Comoros, to Mayotte (8,9). RVF infection was confirmed by IgM serologic analysis. Because of the proximity of Comoros and Mayotte, the RVF situation among humans in Mayotte was assessed. In serum samples from 7 humans with dengue-like syndromes, RVFV IgM or RVFV RNA was detected. We report the isolations and full sequence analysis of 2 RVF viral isolates from these serum specimens.
During January–April 2007, seven patients native to Mayotte were admitted to the hospital for severe dengue-like syndromes. Two patients were RVF seropositive by IgM and IgG, and the other 5 were positive by RVFV-specific reverse transcription PCR (RT-PCR) as detailed in Sissoko et al. (9). As described for other viruses, we used in-house IgM-capture enzyme immunoassays and in-house direct detection for IgG by using microplates coated with RVFV antigen and specific binding by using a peroxidase-labeled goat anti-human IgG conjugate (10).
RVFV isolates were obtained on Vero E6 cells from the serum of 2 hospitalized patients (serum collected on February 21 and March 20, 2008). RNA extracted by using the RNaid Kit (Qbiogene, Carlsbad, CA, USA) was reverse transcribed by PCR and amplified by using SuperScript One-Step RT-PCR with platinum Taq kit (Invitrogen, San Diego, CA, USA) with primers targeting the small, medium, and large segments (adapted from [11]). Overlapping RT-PCR fragments were purified by ultrafiltration. Sequencing reactions were performed by using the Big Dye Terminator v1.1 cycle sequencing kit (Applied Biosystems, Foster City, CA, USA). Sequence chromatograms from both strands were obtained on automated sequence analyzer ABI3730XL (Applied Biosystems). For sequence analysis, contig assemblies and sequence alignments were performed by using BioNumerics version 6.6 (Applied-Maths, Sint-Martens-Latem, Belgium).
We used 2 methods for phylogenetic reconstruction: maximum likelihood and the Bayesian inference. The best models of nucleotide substitution for each dataset were selected from the uncorrected and corrected Akaike Information Criterion, the Hannan and Quinn performance-based decision theory and Bayesian Information Criterion of Jmodeltest version 0.1 and TREEFINDER version October 2008 (Munich, Germany, distributed by its author at www.treefinder.de). The consensus substitution models proposed by the different software packages were selected for further analyses. Comparison of the maximum-likelihood method implemented by the TREEFINDER program with others was performed on the small, medium, and large segments by using the neighbor-joining and maximum parsimony methods from Mega5 software and the Bayesian approach by using MrBayes version 3.0B4 for phylogenetic reconstruction with random starting trees and run for 2,000,000 generations, sampling the Markov chains at intervals of 100 generations (12,13). Branch support values were obtained by using nonparametric bootstrapping with 1,000 resampling for PhyML and TREEFINDER and the posterior probabilities for the Bayesian approach estimated on 10,000 samples (sample frequency set to every 100th generation by using the Markov Chain Monte Carlo sampling). We compared topologies of the maximum-likelihood and Bayesian trees obtained for the different segments.
The complete genome sequences performed on 2 human RVFV isolates from Mayotte referenced as 2008/00099 and 2008/00101 (deposited in GenBank/EMBL under accession nos. HE687302–HE687307) are embedded within the larger 2006–2007 East African clade, specifically within the lineage previously termed Kenya-1 (Table A1). The Kenya-1 virus lineage includes 18 isolates, 8 human isolates (035/07Baringo Kenya 2007, SPU10315 Kenya 2007, Garissa 004/006 Kenya 2006, Dod002/007Tanzania 2007, 3162 Madagascar 2008, 3163 Madagascar 2008, 3164 Madagascar 2008, 3165 Madagascar 2008), 2 mosquito isolates (KLFMsq091/07 Kenya 2007, 131B04/06Garissa Kenya 2006), and 8 livestock isolates (1602Mombassa Kenya 2007, 2820Garissa Kenya 2007, 3644Baringo Kenya 2007, 473Kajaido Kenya 2007, 0611Kenya 2007, 3168 Madagascar 2008, 3169 Madagascar 2008, 3170 Madagascar 2008). The Kenya 2 virus lineage comprises 3 human isolates (1811Garissa Kenya 2006, 0094 Kenya 2007, Tan001/007 Tanzania 2007).
Figure
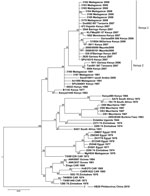
Figure. . . Fifty-two complete sequences of Rift Valley fever virus small genome segments aligned and analyzed by the Bayesian program (MrBayes). Scale bar indicates nucleotide substitutions per site.
Because maximum-likelihood and Bayesian tree topologies obtained for the 3 segments were similar, only the small segment is presented. The Figure shows the Bayesian tree topology based on all RVFV small segments, with the HB29 phlebovirus from the People’s Republic of China as an outgroup. Tree topologies are consistent with those generated in previous work (7,11). The reliability of the phylogenetic trees was confirmed by performing bootstrap analysis. The Kenya 1 and Kenya 2 lineages clustered together with an overall bootstrap value of 92% but with sublineage bootstrap values of 56%–100%.
The work of Sissoko et al. (9) suggested the indigenous transmission of RVFV in humans in Mayotte. The geographic distribution of the 10 human serum samples found positive for RVFV in 2007 and 2008 was not spatially delimited. All case-patients were native to the island and resided in the following districts: Mamoudzou (3), Brandaboua (2), Dembeni (1), Sada (1), Chirongui (2), and Boueni (1). None reported travel into countries where RVF is endemic (9). The genomic analysis of the Mayotte isolates placed them within the 2006–2007 eastern African Kenya-1 lineage. RVF activity in Mayotte appears to be an expansion of the eastern African mainland 2007–2008 outbreak. It illustrates the risk for introduction in Mayotte or other Comorian islands of infectious agents involved in outbreaks in neighboring eastern African coastal countries, the major source being livestock importation from the African mainland or Madagascar.
The recent data published on RVFV Malagasy strains (7,14) support an epidemic cycle with introduction of the virus from outbreaks on mainland eastern Africa rather than an enzootic cycle in Madagascar. RVFV has been isolated from at least 40 species of mosquitoes in 8 genera. Recent experimental RVFV infections on African mosquito species revealed that 8 species—Aedes palpalis (Newstead), Ae. mcintoshi Huang, Ae. circumluteolus (Theobald), Ae. calceatus Edwards, Ae. aegypti (L), Culex antennatus (Becker), Cx. pipiens (L), and Cx. quinquefasciatus Say—are susceptible to infection, and that all except Ae. calceatus, Ae. aegypti, and Cx. quinquefasciatus transmitted RVFV by bite after oral exposure (15). In Mayotte, a preliminary study has shown that 4 species—Ae. circumluteolus, Cx. antennatus, Cx. quinquefasciatus, and Ae. aegypti—are present (T. Balenghien, V. Robert, pers. comm.).
Even if mosquito transmission might have occurred among some of the 7 reported RVF case-patients, contact with imported ruminants is the predominant means of exposure among these reported case-patients. However, further entomologic studies need to be conducted to identify all potential vector species in the island and animal surveys need to be conducted to help detect RVF at early stages to gain a better understanding of the ecologic and climatic factors that favor RVFV dissemination. These assessments will help in the development of appropriate control measures to better predict and respond to potential RVF outbreaks.
Mrs Cêtre-Sossah is a virologist at the Unit Control of Animal and Exotic Diseases at Centre de Coopération Internationale en Recherche Agronomique pour le Développement in Montpellier. Her research interests include the diagnosis and control of communicable and noncommunicable diseases, especially Rift Valley fever, and those caused by poxviruses and bluetongue viruses.
Acknowledgment
We thank Vincent Michaud for his steadfast support.
References
- Daubney R, Hudson JR, Garnham PC. Enzootic hepatitis or RVF. An undescribed virus disease of sheep cattle and man from East Africa. J Pathol Bacteriol. 1931;34:545–79. DOIGoogle Scholar
- Shoemaker T, Boulianne C, Vincet MJ, Pezzanite L, Al-Qahtani MM, Al-Mazrou Y, Genetic analysis of viruses associated with emergence of Rift Valley fever in Saudi Arabia and Yemen, 2000–2001. Emerg Infect Dis. 2002;8:1415–20.PubMedGoogle Scholar
- Schmaljohn CS, Nichol ST. Bunyaviridae. In: Knipe DM, Howley PM, editors. Fields virology, 5th ed. Philadelphia: Lippincott, Williams & Wilkins; 2007. p. 1741–89.
- McIntosh BM, Russell D, Dos Santos I, Gear JH. Rift Valley fever in humans in South Africa. S Afr Med J. 1980;58:803–6.PubMedGoogle Scholar
- Meegan JM, Watten RH, Laughlin LW. Clinical expeience with Rift valley fever in humans during the 1977 Egyptian epizootic. Contribution to Epidemiology and Biostatistics. 1981;3:114–23.
- Bird BH, Githinji JW, Macharia JM, Kasiiti JL, Muriithi RM, Gacheru SG, Multiple virus lineages sharing recent common ancestry were associated with a large Rift Valley fever outbreak among livestock in Kenya during 2006–2007. J Virol. 2008;82:11152–66. DOIPubMedGoogle Scholar
- Andriamandimby SF, Randrianarivo-Solofoniaina AE, Jeanmaire EM, Ravololomanana L, Razafimanantsoa LT, Rakotojoelinandrasana T, Rift Valley fever during rainy seasons, Madagascar, 2008 and 2009. Emerg Infect Dis. 2010;16:963–70.PubMedGoogle Scholar
- ProMED-mail. Comoros, RFI- Rift Valley fever, human, bovine. ProMED-mail. 2007 Nov 27 [cited 2007 Nov 29]. http://www.promedmail.org, article no. 20071129.3855.
- Sissoko D, Giry C, Gabrie P, Tarantola A, Pettinelli F, Collet L. Rift Valley fever, Mayotte, 2007–2008. Emerg Infect Dis. 2009;15:568–70. DOIPubMedGoogle Scholar
- Del Giudice P, Schuffenecker I, Vandenbos F, Evelyne C, Zeller H. Human West Nile virus, France. Emerg Infect Dis. 2004;10:1885–6. DOIPubMedGoogle Scholar
- Bird BH, Khristova ML, Rollin PE, Ksiazek TG, Nichol ST. Complete genome analysis of 33 ecologically and biologically diverse Rift Valley fever virus strains reveals widespread virus movement and low genetic diversity due to recent common ancestry. J Virol. 2007;81:2805–16. DOIPubMedGoogle Scholar
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9. DOIPubMedGoogle Scholar
- Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–5. DOIPubMedGoogle Scholar
- Carroll SA, Reynes JM, Khristova ML, Andriamandimby SF, Rollin PE, Nichol ST. Genetic evidence for Rift Valley fever outbreaks in Madagascar resulting from virus introductions from the East African mainland rather than enzootic maintenance. J Virol. 2011;85:6162–7. DOIPubMedGoogle Scholar
- Turell MJ, Linthicum KJ, Patrican LA. Vector competence of selected African mosquito (Diptera: Culicidae) species for Rift Valley fever virus. J Med Entomol. 2008;45:102–8. DOIPubMedGoogle Scholar
Figure
Table
Cite This ArticleTable of Contents – Volume 18, Number 6—June 2012
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Catherine Cêtre-Sossah, CIRAD, Département Bios, UMR15, TA A-15/G, Campus International de Baillarguet, F-34398 Montpellier Cedex 5, France
Top