Volume 22, Number 1—January 2016
Dispatch
Variations in Spike Glycoprotein Gene of MERS-CoV, South Korea, 2015
Cite This Article
Citation for Media
Abstract
An outbreak of nosocomial infections with Middle East respiratory syndrome coronavirus occurred in South Korea in May 2015. Spike glycoprotein genes of virus strains from South Korea were closely related to those of strains from Riyadh, Saudi Arabia. However, virus strains from South Korea showed strain-specific variations.
Since it was first identified in 2012, Middle East respiratory syndrome coronavirus (MERS-CoV) has emerged as a novel viral pathogen that causes severe acute respiratory illness, including fever, cough, and shortness of breath (1). The current outbreak of infection with this virus in South Korea, which began on May 20, 2015, has infected 186 patients and caused 36 deaths within 2 months. This developing public health concern has attracted worldwide attention as a potential cause of a global pandemic. Although extensive biologic and clinical characterization should be performed to measure the public health effect of this outbreak, currently available genetic data are informative in clarifying virus alterations that affect transmissibility.
MERS-CoV spike (S) glycoprotein binds cellular receptor dipeptidyl peptidase 4 (DPP4, CD26) for host cell entry (2), and a receptor-binding domain (RBD) on virus S protein mediates this interaction (3). In addition, S proteins expressed on the virus surface can induce host antibodies that block MERS-CoV entry (4). To investigate changes in the S gene associated with viral evolution and possible genetic markers of altered transmissibility, an analysis of S genes obtained from clinical specimens during the early phase of the outbreak was performed.
We identified genetic variability of MERS-CoV S genes among infected persons in South Korea. Samples from 7 patients identified as positive for MERS-CoV were sequenced. These patients were identified by using sequences upstream of the envelope protein gene and open reading frame (ORF) 1a in real-time reverse transcription PCRs (5) (Table 1).
Figure 1

Figure 1. History of confirmed cases of Middle East respiratory syndrome coronavirus infection, South Korea, May 2015. Eight confirmed cases of human infection with this virus are shown according to date of onset...
Index case-patient 1 (PAT001) had traveled to Bahrain, the United Arab Emirates, and Saudi Arabia during April 24–May 4, 2015, and became symptomatic on May 11 after his return to South Korea (6). After he visited a local clinic, his symptoms worsened, and he was hospitalized on May 15. During his hospitalization (May 15–17), PAT001 shared a room with PAT003 and the same ward with PAT009, PAT012, PAT013, and PAT015. PAT042 was admitted to the same hospital on May 19 (Figure 1). PAT010 was the son of PAT003 and had visited his father in the hospital before traveling to China, where he became symptomatic and tested positive for MERS-CoV. PAT002 was the wife of PAT001 and cared for him during his illness.
The S gene was amplified directly from nucleic acids extracted from respiratory specimens (6 patients) or a viral isolate (1 patient) (7) by using the QIAamp Viral RNA Mini Kit (QIAGEN, Hilden, Germany). Reverse transcription was performed by using the Superscript III First-Strand Synthesis System (Life Technologies, Bleiswijk, the Netherlands) and virus-specific reverse primers. cDNA was amplified by using an overlapping PCR to generate products of 600–3,000 bp that covered the entire S gene.
Resulting PCR amplicons were sequenced by using Sanger sequencing with an ABI 3730 Analyzer (Applied Biosystems, Foster City, CA, USA) or next-generation sequencing. For next-generation sequencing, PCR products were pooled and fragmented to an average of 300 bp, and a sequencing library was constructed by using the Illumina TruSeq Nano DNA Sample Prep Kit (Illumina, San Diego, CA, USA). Sequencing was performed by using the Illumina MiSeq Platform (Illumina).
To explore relationships of newly isolated virus strains from South Korea with other MERS-CoV strains, 131 reference MERS-CoV S gene sequences from GenBank and MERS-CoV Sequences June 2015 (http://tinyurl.com/MERS-CoV-4Jun15) (8) and 8 strains from South Korea, including a sequence from PAT010 (ChinaGD01; Chinese Centers for Disease Control and Prevention, Beijing, China), were aligned by using MUSCLE software (9). This alignment was used for subsequent phylogenetic analysis. A phylogenetic tree was constructed by using the maximum-likelihood method with a bootstrap value of 1,000 and RAxML software (10).
Figure 2
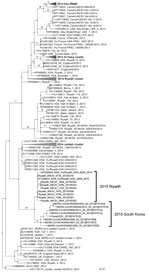
Figure 2. Molecular phylogenetic tree and coding region variants for spike glycoprotein genes of Middle East respiratory syndrome coronavirus (MERS-CoV) isolates from South Korea, May 2015, and reference MERS-CoV sequences. Phylogenetic analysis of...
All 8 ORFs from virus isolates obtained during the outbreak in South Korea were most closely related to ORFs of the recently isolated 2015 Riyadh clade, but isolates from South Korea constituted a novel branch, which was supported by a bootstrap value of 87% (Figure 2). Phylogenetic data indicated that virus isolates from other patients originated from virus isolates from the index case-patient. These data also showed that strains detected in 2015 formed 2 groups: KSA-2466–like viruses and KKUH_0734–like viruses. Viruses from South Korea isolated in 2015 clustered with 1 sublineage of KKUH_0734–like viruses from Saudi Arabia.
Nucleotide sequence comparisons with 131 reference MERS-CoV S genes showed that the clade from South Korea had highest identity (99.68%–99.9%) with recently circulating strains from Riyadh isolated in 2015. Strains from South Korea had 8 novel nucleotide substitutions (C183G, A409C, T1586C, G1588C, T1848C, G1886A, T3177C, and C3267T) that are unique to the South Korea lineage and share nucleotide substitution T258C with some viruses from Saudi Arabia detected earlier in 2015 (Table 2). T3177C and C3267T mutations were observed only in all viruses from South Korea.
Of the 8 nucleotide substitutions, 4 (A409C, T1586C, G1588C, and G1886A) were nonsynonymous and resulted in 4 amino acid changes (S137R, I529T, V530L, and R629H) (Table 2). Among these mutations, 2 nonsynonymous variants (S137R and V530L) were identified in isolates from PAT002 after the third passage in Vero cells and were assumed to be cell culture–adaptive mutations (11). The I529T and V530L mutations were located in the RBD, but not at the RBD–DPP4 receptor interface (3). The R629H mutation was situated outside the RBD. However, on the basis of only these results, we could not determine whether these amino acid substitutions affected receptor-binding affinity between human DPP4 receptor and MERS-CoV S protein.
To understand the rate at which virus genetic diversification occurred during the outbreak in South Korea, we used the Bayesian–Markov Chain Monte Carlo method in BEAST version 2.1.3 (http://beast2.org/) for 8 S genes. The Hasegawa, Kishino, and Yano substitution model was selected under uncorrelated lognormal molecular clock and a birth–death coalescent.
The S gene was estimated to evolve at mean rate of 6.72 × 10−3 substitutions/site/year (95% highest posterior density [HPD] 5.59−6.93 × 10−3 substitutions/site/year). This mutation rate for the S gene was higher than that for complete MERV-CoV genomes in other studies: 1.12 × 10−3 substitutions/site/year (95% HPD 8.76 × 10−4−1.37 × 10−3 substitutions/site/year) (12) and 9.29 × 10−4 substitutions/site/year (95% HPD 7.19 × 10−4−1.15 × 10−3 substitutions/site/year) (13). However, more data are required to demonstrate the pattern of MERS-CoV evolution during the outbreak in South Korea because results are limited by a relatively low number of sequences, short selected time points, examination of only the S gene region, and different sequencing methods.
Accurate genome sequencing can identify spatiotemporal patterns that help understand dynamics of rapid spread of MERS-CoV infection. We report S glycoprotein gene sequences of MERS-CoV from 8 patients and a strain cultured in Vero cells. Genetic information obtained is useful for understanding the evolutionary history of MERS-CoV.
On the basis of our phylogenetic analyses, virus sequences of strains isolated in South Korea in 2015 form a unique clade. Genetic variations elucidated in this study show an unreported sequence in the RBD, which suggests that MERS-CoV circulating in South Korea during the outbreak in 2015 has higher genetic variability and mutation rates. However, we cannot conclude that deleterious effects promoting spread of infection will occur because of these mutations. Additional genetic information will resolve precise characteristics of the MERS-CoV obtained during the outbreak in South Korea.
Dr. Dae-Won Kim is a principle researcher at the Korea National Institute of Health, Korea Centers for Disease Control and Prevention, Cheongju-si, South Korea. His research interests include biology of virus systems, comparative genomics of pathogens, and immunoinformatics.
Acknowledgments
We thank Malik Peiris for informative discussions.
This study was supported by grants from the Intramural Research Fund of the Korea National Institute of Health (no. 2014-N47001-00) and the Intramural Fund of the Korea Centers for Disease Control and Prevention (nos. 4834-300-210-13 and 4837-301-210-13).
References
- Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367:1814–20 and.PubMedGoogle Scholar
- Raj VS, Mou H, Smits SL, Dekkers DH, Muller MA, Dijkman R, Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature. 2013;495:251–4 and.PubMedGoogle Scholar
- Wang N, Shi X, Jiang L, Zhang S, Wang D, Tong P, Structure of MERS-CoV spike receptor-binding domain complexed with human receptor DPP4. Cell Res. 2013;23:986–93 and.PubMedGoogle Scholar
- Song F, Fux R, Provacia LB, Volz A, Eickmann M, Becker S, Middle East respiratory syndrome coronavirus spike protein delivered by modified vaccinia virus Ankara efficiently induces virus-neutralizing antibodies. J Virol. 2013;87:11950–4 and.PubMedGoogle Scholar
- World Health Organization WHO). Laboratory testing for Middle East respiratory syndrome coronavirus. Interim guidance (revised). WHO/MERS/LAB/15.1. Geneva: The Organization; 2015 [cited 2015 Oct 15]. http://apps.who.int/iris/bitstream/10665/176982/1/WHO_MERS_LAB_15.1_eng.pdf
- World Health Organization Regional Office for the Western Pacific (WHO/Western Pacific Region). Middle East respiratory syndrome coronavirus (MERS-CoV)—Republic of Korea. Manila: WHO/Western Pacific Region; May 30, 2015 [cited 2015 Oct 5]. http://www.who.int/csr/don/30-may-2015-mers-korea/en/
- Kim YJ, Cho YJ, Kim DW, Yang JS, Kim H, Park S, Complete genome sequence of Middle East respiratory syndrome coronavirus KOR/KNIH/002_05_2015, isolated in South Korea. Genome Announc. 2015;3:e00787–15.PubMedGoogle Scholar
- Somily A, Barry M, Al Subaie SS. BinSaeed AA, Alzamil FA, Zaher W, et al. 2015. Evolution patterns of the Middle East respiratory syndrome coronavirus (MERS-CoV) obtained from MERS patients in early 2015 [cited 2015 Oct 5]. http://virological.org/uploads/default/original/1X/40ca96d0d6ceb95bacbda00b974b50170d412d9b.pdf
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7 and.
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3 and.
- Mohapatra JK, Pandey LK, Rai DK, Das B, Rodriguez LL, Rout M, Cell culture adaptation mutations in foot-and-mouth disease virus serotype A capsid proteins: implications for receptor interactions. J Gen Virol. 2015;96:553–64 and.
- Cotten M, Watson SJ, Zumla AI, Makhdoom HQ, Palser AL, Ong SH, Spread, circulation, and evolution of the Middle East respiratory syndrome coronavirus. MBio. 2014;5:e01062–13 and.
- Barry MA, Al Subaie SS, Somily AM. BinSaeed AA, Alzamil FA, Al-Jahdali IA, et al. Recent evolution patterns of Middle East respiratory syndrome coronavirus (MERS-CoV), 2015 [cited 2015 Oct 15]. http://virological.org/uploads/default/78/4a6e0a6e90d92867.pdf
Figures
Tables
Cite This Article1These authors contributed equally to this article.
Table of Contents – Volume 22, Number 1—January 2016
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Sung Soon Kim, Division of Respiratory Viruses, Center for Infectious Diseases, Korea National Institute of Health, Korea Centers for Disease Control and Prevention, Cheongju-si, South Korea
Top