Volume 23, Number 1—January 2017
Dispatch
Host-Associated Absence of Human Puumala Virus Infections in Northern and Eastern Germany
Figure 2
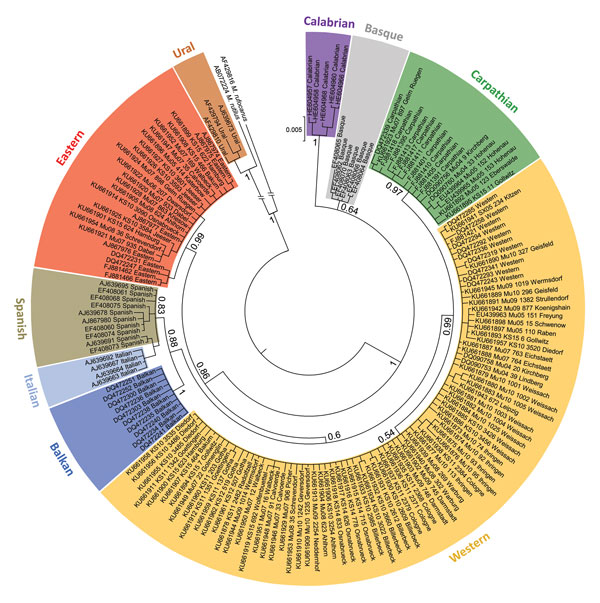
Figure 2. Phylogenetic relationships of European bank vole lineages. Sequences are categorized on the basis of mitochondrial cytochrome b gene sequences and shown as a maximum clade credibility phylogenetic tree with posterior probabilities displayed for major nodes. Novel sequences are labeled with individual code and trapping site (Technical Appendix Table). Additional published sequences are included as references for bank vole evolutionary lineages, labeled with GenBank accession number followed by lineage indication. Phylogenetic analyses were performed with MrBayes version 3.2.2 (https://sourceforge.net/projects/mrbayes/files/mrbayes/) on the CIPRES platform for 166 cytochrome b sequences of 843-bp length. A mixed nucleotide substitution matrix was specified in 4 independent runs of 107 generations for the data set. A burn-in fraction of 25% was discarded and samples were recorded every 103 generations. Cytochrome b sequences of M. rutilus and M. rufocanus voles were used as outgroups.
1Current affiliation: Seramun Diagnostica GmbH, Heidesee, Germany.
2Current affiliation: Stiftung Tierärztliche Hochschule Hannover, Hannover, Germany.