Volume 23, Number 2—February 2017
Research
Spread and Evolution of Respiratory Syncytial Virus A Genotype ON1, Coastal Kenya, 2010–2015
Cite This Article
Citation for Media
Abstract
In February 2012, the novel respiratory syncytial virus (RSV) group A, genotype ON1, was detected in Kilifi County, coastal Kenya. ON1 is characterized by a 72-nt duplication within the highly variable G gene (encoding the immunogenic attachment surface protein). Cases were diagnosed through surveillance of pneumonia in children at the county hospital. Analysis of epidemiologic, clinical, and sequence data of RSV-A viruses detected over 5 RSV seasons (2010/2011 to 2014/2015) indicated the following: 1) replacement of previously circulating genotype GA2 by ON1, 2) an abrupt expansion in the number of ON1 variants detected in the 2014/2015 epidemic, 3) recent accumulation of amino acid substitutions within the ON1 duplicated sequence, and 4) no clear evidence of altered pathogenicity relative to GA2. The study demonstrates the public health importance of molecular surveillance in defining the spread, clinical effects, and evolution of novel respiratory virus variants.
Respiratory syncytial virus (RSV) is a major cause of pneumonia and bronchiolitis among infants and children globally (1,2). Although immune responses develop in those who have had RSV infection during childhood, these persons remain susceptible to RSV upper respiratory tract reinfection throughout life (3). No licensed RSV vaccine exists. Of the 11 proteins encoded by the RSV genome, the attachment glycoprotein (G) is the most variable and has been shown to accumulate amino acid changes over time (4). RSV is classified into 2 groups, RSV-A and RSV-B (5); each group is divided into genotypes (6), and these are further characterized into variants (7). Globally, RSV viruses belonging to different groups, genotypes, and variants often co-circulate in epidemics (7,8). The phenomenon of reinfection and difficulty in developing a vaccine may in part be due to the antigenic diversity and variability in the virus (9).
Two novel RSV genotypes with large duplications of amino acids in the attachment G glycoprotein have been detected globally. In 1999, the BA genotype was detected in Buenos Aires, Argentina; the genotype had a 60-nt duplication within the C-terminal region of the G gene (10). The BA variant subsequently spread rapidly throughout the world, becoming the predominant group B genotype, and in some regions replacing all previous circulating RSV-B genotypes (11). More recently, in December 2010, genotype ON1, with a 72-nt duplication (also within the C-terminal region of the G gene), was detected in Ontario, Canada (12). Viruses belonging to this genotype have rapidly spread and diversified globally (13–20). Such emergent genotypes appear to have a fitness advantage over preceding genotypes of the same RSV group (21). Of public health interest is whether increased fitness is associated with increased severity and immune evasion (with potential vaccine modality implications).
The temporal progression of RSV genotypes can be followed directly because of the unique tags (the duplications), which provides a rare opportunity to learn more about the introduction, spread, severity, and related selection processes (including immune evasion) for RSV and to obtain insights into the nature of emergence of novel virus variants. In this regard, we undertook an in-depth analysis of RSV-A genotype ON1 epidemiology in Kilifi, a county in coastal Kenya. In Kilifi, RSV epidemics typically begin during September–November of 1 year and continue until July–August of the following year, with a peak in cases during January–March. We have analyzed sequence data collected over 5 RSV epidemic seasons in Kilifi (2010/2011 to 2014/2015), which includes the period after the initial detection of this novel genotype within Kilifi.
Study Location and Population
The study was undertaken in Kilifi County and is part of surveillance aimed at understanding the epidemiology and disease effects of RSV-associated pneumonia cases in this region (22). Respiratory swab samples (combined nasopharyngeal and oropharyngeal) were collected from September 2010 through August 2015 from children ages 1 day to <5 years admitted to Kilifi County Hospital (KCH) with syndromically defined severe or very severe pneumonia (referred to here as lower respiratory tract infections, LRTIs), as defined in Table 1 and previously (22)
Study Samples and Laboratory Procedures
All specimens were screened for RSV by 2 methods (22–24). Raw samples were tested for RSV antigen by immunofluorescence antibody test (Chemicon International Inc., Temecula, CA, USA). Viral RNA was extracted from respiratory samples using QIAamp Viral RNA Mini Kit (QIAGEN, Hilden, Germany) and tested for RSV (differentiating groups A and B) by multiplex real-time reverse transcription PCR. All RSV-positive samples by either test were taken forward for processing. In addition, a small number of RSV-negative samples were sequenced.
The viral RNA was reverse transcribed into cDNA by using the Omniscript RT Kit (QIAGEN). The cDNA was then amplified with primers targeting the G ectodomain region (3,25), and the amplicons were sequenced by using BigDye version 3.1 chemistry on an ABI 3130xl (Applied Biosystems, Waltham, MA, USA). Sequence reads were assembled into contigs by using Sequencher version 5.0.1 (Gene Codes Corp., Ann Arbor, MI, USA). The sequences analyzed here have been deposited in GenBank (accession nos. KX453303–KX453534); previously reported sequences from Kilifi added to this analysis had also been deposited in GenBank (accession nos. KF587911–KF588014) (13).
Global Comparison Dataset
To determine the relatedness of the Kilifi viruses to those circulating around the world and thereby clarify their global context, we downloaded all RSV-A G-gene sequences collected during 2010–2015; deposited in GenBank as of January 19, 2016; and 241 nt to 687 nt in length. A total of 995 sequences from 24 countries were used in this analysis. For the whole dataset and for some countries, we further grouped sequences by calendar year for temporal analysis. We subsampled unique sequences (sequences that differed by >1 nt from any other sequence over the sequenced region) by epidemic season (Kilifi only) or per calendar year.
Sequence Alignments and Diversity Analysis
All sequences, from Kilifi and the global dataset, were collated and aligned using MAFFT (multiple alignment using fast Fourier transform) alignment software version 7.272 (26). We calculated the variability of nucleotides and amino acids using MEGA 6.06 (27).
Phylogenetic Analyses
We used MEGA 6.06 to infer maximum-likelihood phylogenetic trees under the general time reversible model with the site heterogeneity gamma model (27). This model was the best substitution model as determined by IQ-TREE version 1.4.2 (28). Bootstrapping with 1,000 iterations was implemented to evaluate branch support of the phylogenetic clusters. We assigned RSV-A genotypes as previously determined by Peret et al. (6) and Eshaghi et al. (12). To position the genotype ON1 viruses in the global context, we examined ON1 lineages as recently assigned by Duvvuri et al. (20).
RSV-A Variants Analysis
We determined the number of genotype GA2 and ON1 variants circulating in Kilifi and globally using a recently developed pragmatic criterion (7,8). In brief, a variant is a virus or a group of viruses within a genotype that possesses ≥4 nt differences in the G ectodomain region when compared with other viruses. This analysis was done using usearch v8.1.1861 (29).
Protein Substitution and Selection Analysis
The N-glycosylation sites were predicted by using the NetNGlyc 1.0 server (30). We only considered the default Asn-X-Ser/Thr sequon (when X was not proline) for prediction. We also analyzed for patterns of change in amino acids using python scripts. Finally, we looked for potential positively selected and co-evolving sites using the Datamonkey server (http://www.datamonkey.org/). For positive selection analysis, we used 3 methods: SLAC (single likelihood ancestor counting), FEL (fixed effects likelihood), and MEME (mixed effects model for evolution).
Statistical Analyses
We explored associations between demographic, clinical, or outcome variables and RSV genotypes for all cases of RSV-positive severe and very severe pneumonia. We used logistic regression computing odds ratios (ORs) in Stata version 13 (StataCorp LP, College Station, TX, USA).
Over the 5 RSV epidemics examined (2010/2011 to 2014/2015), a total of 4,010 samples were collected from eligible children; 3,561 (88.8%) were tested for RSV and 881 (24.7%) RSV-positive samples were identified (Technical Appendix Table 1). Of these samples, 600 (68.1%) were RSV-A. The G gene was successfully sequenced in 442 (73.7%) samples. An additional 41 sequences were available from samples that were negative by both immunofluorescent antibody test and PCR or from patients with mild pneumonia (data for these cases were not included in the clinical severity analysis). Thus, we carried 483 sequences for phylogenetic analysis. The sequences ranged from 618 nt to 690 nt in length, corresponding to nucleotides 295–912 of the reference strain A2 (M74568).
Figure 1
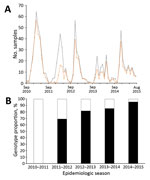
Figure 1. Circulating patterns of respiratory syncytial virus (RSV) in Kilifi, Kenya, September 2010– August 2015. A) Total RSV-positive cases (gray continuous line) and typed RSV-A samples (dotted orange line) by month. B)...
We found that 2 RSV-A genotypes were circulating in Kilifi: ON1 (n = 283, 58.6%) and GA2 (n = 200, 41.4%). The temporal prevalence of the total RSV, RSV-A, and genotypes ON1 and GA2 is shown in Figure 1 and Technical Appendix Table 1. We observed rapid replacement of the previously circulating dominant GA2 genotype by ON1 in Kilifi, from a prevalence of 0% in the 2010/2011 epidemic to a prevalence of 67.4% in 2011/2012 when ON1 was first detected in Kilifi, and to 96.1% in the recent 2014/2015 epidemic. In addition, RSV-A predominated in 3 consecutive RSV epidemics from 2012/2013 to 2014/2015.
To investigate the demographic and clinical effects of RSV-A genotype ON1 in Kilifi, we compared the proportions of GA2- and ON1-infected case-patients by sex, age, clinical features of cough, difficulty in breathing, chest wall indrawing, inability to drink, hypoxia, prostration/consciousness, pneumonia status (severe or very severe pneumonia), length of hospital stay, and death at the hospital (Table 1). The proportions of both genotypes were very similar for the demographic and clinical characteristics analyzed. However, the proportion of patients with ON1 infection who were unable to eat was more than double that for GA2-infected case-patients (18.9% vs. 8.8%), and this difference was found significant by logistic regression (OR 2.40, 95% CI 1.31–4.36; Table 2). Nonetheless, the proportion with very severe pneumonia was no higher in ON1 infections than in GA2 infections (OR 0.89, 95% CI 0.57–1.39).
Figure 2
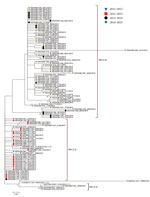
Figure 2. Maximum-likelihood phylogenetic tree of unique respiratory syncytial virus (RSV) genotype ON1 G gene ectodomain sequences from Kilifi, Kenya, 2012–2015. The taxa are color coded by the epidemic season of detection (key),...
The maximum-likelhood tree shows the clustering of unique genotype ON1 sequences in Kilifi (Figure 2). Two genotype ON1 lineages recently defined by Duvvuri et al. (20) are shown to be circulating in Kilifi: lineages ON1 [1.1] and ON1 [1.3]. Of these 2 lineages, ON1 [1.3] was the most prevalent in 2011/2012 and 2012/2013. However, a potential new lineage, denoted here as ON1 [1.4], clustered away from ON1 [1.3] and seemed to have recently arisen comprising sequences from the strains circulating in the 2013/2014 and 2014/2015 epidemics. The genetic divergence (p distance) between ON1 [1.4] and the other ON1 lineages identified in Kilifi ranged from 0.013 to 0.045, similar to the genetic distances between the previously defined ON1 lineages.
We detected a total of 66 RSV-A variants during the entire surveillance period in Kilifi (Table 3). The variants comprised 1–82 sequences; 39 (59.1%) of the 66 variants were singletons. Most variants did not persist between epidemics (46/66 [69.7%]). However, 14 variants persisted for 2 consecutive seasons, 1 for 4 consecutive seasons, and 5 for 2 nonconsecutive seasons. Therefore, the number of variants (accumulated by epidemic) increased to 86 by 2014/2015. The number of GA2 variants declined consistently, from 17 variants in 2010/2011 (before ON1 arrived) to only 4 variants in 2014/2015. On the other hand, the number of ON1 variants assigned remained at 5 variants between 2011/2012 and 2012/2013 before increasing to 8 variants in 2013/2014, then rising markedly to 25 variants in 2014/2015.
Seven codon sites were predicted to be N-glycosylated within the G protein for the Kilifi sequences: 4 sites for genotype ON1 viruses (codons 103, 135, 237, 318) and 6 sites for genotype GA2 viruses (codons 103, 135, 237, 251, 273, 294). However, none of the potential N-glycosylation sites occurred within the 72-nt duplication of the ON1 viruses. Notably for GA2 viruses, sites 237 and 273 were mutually exclusive: a virus belonging to this genotype had either these sites potentially N-glycosylated but not both (Technical Appendix Figure 1).
Figure 3

Figure 3. Amino acid substitutions in respiratory syncytial virus A (RSV-A) G protein for sequences isolated in Kilifi Kenya from season 2011/2012 to 2014/2015. All unique protein sequences per epidemic were collated, aligned...
The nucleotide variability and amino acid variability over the 4 seasons are shown in Technical Appendix Table 2. Amino acid substitutions over the sequenced portion of the G protein are shown in Figure 3, panel A. Two codon positions possessed amino acid substitutions that distinguished between ON1 (232G, 253K) and GA2 (232E, 253T) viruses. In addition, the new ON1 [1.4] lineage viruses seem to have fixed a threonine (I/T136T) and acquired a unique substitution (P206Q) that distinguishes them from the other ON1 lineages.
Figure 3, panels B and C, illustrates amino acid substitutions within the duplication region of the Kilifi ON1 viruses. We designated the first set of 72 nt as duplication sequence I and the second set as duplication sequence II. Within this region, we observed that over the 3 seasonal epidemics from 2011/2012 to 2013/2014 and early (September–November) in the 2014/2015 epidemic, amino acid substitutions only occurred within the duplicated sequence I except for 3 substitutions in 2 viruses within the duplicated sequence II. Beginning in December 2014, however, we found numerous substitutions in duplicated sequence II with 2 adjacent and corresponding positions between the duplicated sequences I and II acquiring similar amino acid substitutions (i.e., Y273H and Y297H, P274L/S and P298L/R). Furthermore, sites 273 and 297 were detected to be co-evolving from the Spidermonkey analysis (http://www.datamonkey.org/). However, only 1 ON1 codon site (251) was identified to be positively selected with p<0.05 by >1 method (SLAC, FEL, or MEME).
Using global datasets for genotypes ON1 and BA, we compared the temporal detection of RSV variants within each of these genotypes during the first 5 and 10 years, respectively, from initial detection (Technical Appendix Table 3). We observed an explosion of new ON1 variants globally, from 8 variants in 2011, to 78 variants in 2012, to 153 variants in 2013. However, the number of ON1 variants decreased in 2014 and 2015, which corresponded with a substantial decrease in both the number of ON1 sequences available in GenBank and the countries that have deposited sequences from these years. On the other hand, the number of BA variants seemed to follow a stepwise or punctuated pattern, whereby the number of variants was stable at 1–6 during 1996–2001, dramatically increased and stabilized at 20–30 variants during 2002–2004, and again sharply rose to 82 variants in 2005 (Technical Appendix Table 3). At the country level (Technical Appendix Table 4), the rapid rise in the number of ON1 variants detected in Kilifi was also observed in the Philippines and Germany, although the number of BA variants detected in some of the countries sampled remained relatively stable over time.
We provide a detailed analysis on the spread and the associated demographic, clinical, and evolutionary characteristics of the novel RSV-A genotype ON1 in Kilifi. ON1 was first detected in Kilifi in February 2012, and within that RSV epidemic (2011/2012), it displaced the previous dominant genotype GA2 by attaining a prevalence of 67%. Its dominance has continued to rise to 96% within a span of 4 epidemics. This rapid rate of replacement is unlike previous replacement rates in the same location; for example, when GA5 was displaced by GA2, it took GA2 ≈7 years to reach a prevalence of 95% (8). ON1 seems to possess a fitness advantage over GA2. If such fitness is the result of immune evasion, this characteristic has potential implications for vaccines to deliver population level immunity by herd protection (31).
We found evidence that infections caused by RSV ON1 are more severe than those caused by GA2, showing a higher prevalence of patients’ inability to eat. However, overall numbers of cases of very severe pneumonia were equal for both genotypes. The data, therefore, do not provide a strong indication of more severe disease arising from the ON1 variant. Duvvuri et al. (20) reported significant association of RSV ON1 infection with female patients, which was not evident in our study. Yoshihara et al. (32) reported that cases of upper respiratory infection caused by RSV ON1 in Vietnam caused were significantly associated with clinically severe manifestations of wheezing, tachypnea, and difficulty in breathing compared to infections caused by RSV NA1, whereas Panayiotou et al. (33), on the contrary, reported that children infected with RSV ON1 in Cyprus experienced significantly milder illness than those infected with RSV GA2. Some studies have reported no differences at all between infections with these genotypes. The discordant results may arise from methodologic differences in analyses, clinical disease definitions, and study designs, chance effects resultant from inadequate sample sizes, differences between viruses in different locations, or even host/environmental differences. Prospective studies specifically designed to evaluate virulence or clinical differences between genotypes may offer more reliable insight.
We noted a change in the alternation of RSV subgroup dominance pattern in Kilifi since ON1 was introduced into this community. While ON2 was replacing GA2, it also appeared to also exclude group B strains. RSV-A predominated over RSV-B in 3 consecutive epidemics from 2012/2013 to 2014/2015. Previously, according to data collected during 2002–2012 in Kilifi, RSV-A predominated in up to 2 consecutive epidemics (8). However, it is unclear whether this is a direct effect of ON1, a general change in RSV epidemiologic patterns, or a chance occurrence.
Globally, the prevalence of ON1 seems to vary by location. In Ontario, Canada, where ON1 was first detected in December 2010, the prevalence of ON1 has remained stable at 11%–13% (20). Other countries that have similarly reported ON1 prevalence rates <20% include South Africa (34) and China (35). Reports from Italy (14), South Korea (36), United States (17), Malaysia (37), Japan (19), Thailand (16), Latvia (38), and Cyprus (33) indicate varied RSV ON1 prevalence of 20%–70%. A recent article reported that ON1 was the sole (100%) RSV-A genotype in Buenos Aires, Argentina, in 2014 (39). The varying prevalence suggests that even though ON1 is rapidly spreading globally, host or ecologic differences may determine RSV spread. However, the conclusions of host and ecological differences potentially driving varying prevalence rates in different countries may have been confounded by inadequate/short surveillance periods in these countries.
As shown here and in previous research from Kilifi (7,8), RSV epidemics are composed of multiple variants, each differing sufficiently to suggest separate introductions into the community (as opposed to arising from diversification during the epidemic). In addition, these variants often do not persist between epidemics, which suggests that each year 1) variants generate local herd immunity, leading to their demise, thus requiring reintroductions; or 2) that many invading variants compete in seeding new seasonal epidemics, and that the preceding year variants lose out (perhaps on a chance basis or as stated above because they are less fit due to variant specific immunity).
RSV accumulates amino acid changes over time (4), and we have similarly observed accumulation of amino acid changes in the Kilifi ON1 viruses. It is of interest that in the first 3 RSV epidemic seasons in which we detected ON1 in Kilifi, amino acid substitutions were almost always restricted to the duplicated sequence I of the 72-nt duplication. However, in the 2014/2015 epidemic a virtual explosion in amino acid substitutions was observed within the duplicated sequence II that coincided with a surge in the number of detected ON1 variants. In addition, similar amino acid substitutions occurred in 2 adjacent and corresponding sites in the duplicated sequences I and II, with 1 set of these sites co-evolving. The longer attachment protein of the 72-nt duplication in ON1 viruses appears to offer more opportunities for variable changes and thus greater diversity and increased fitness over previous group A genotypes.
Two codon sites, 232 and 253, within the G protein region analyzed were found to distinguish between genotype ON1 and GA2 viruses. The amino acid change Glu-232-Val has been reported for RSV-A escape mutants that result in loss of reactivity to a specific monoclonal antibody (4). Furthermore, a functional analysis of the 60-nt duplication in BA strains has shown that the duplicated region in the G protein of these viruses augment their fitness (21). While a similar analysis has not been reported for ON1, it is plausible that the increased fitness observed in ON1 is largely due to the 72-nt duplication. However, G-protein N-glycosylation seems to play no role in the increased fitness of ON1 as similar potential N-glycosylation codon sites were detected in both ON1 and GA2 and no additional N-glycosylation sites were detected within the ON1 duplication region.
The rapid diversification of ON1 observed in Kilifi seems to reflect rapid expansion at the global level. While sampling variability may play a role, and similar to varying prevalence, there was variability in the diversification of ON1 viruses in different countries. The number of ON1 variants seemed stable in some countries (e.g., Japan) while expanding in others (e.g., Germany and Philippines). The temporal distribution of BA variants in different countries, however, was mostly stable. Comparisons in the temporal patterns of genotypes BA and ON1 variants may highlight differences between the RSV group B and A viruses. Increased sampling and surveillance will help illuminate on whether such inter-genotypic and RSV group differences are due to ecologic differences or variable sampling.
In conclusion, it is evident that genotype ON1 is not only rapidly spreading globally but also fast evolving. The result is the near exclusion of the previous dominant group A GA2 genotype. The implications of this apparent increased fitness of RSV-ON1 have yet to be resolved. There is some evidence for increased severity of the virus but this is by no means clear or consistent across studies. Continued surveillance for cases together with collection of detailed standardized clinical data are warranted. The possibility exists that ON1 and other similar new RSV variants (e.g., the BA genotype) gain dominance by evading host immunity. It is reasonable to assume this could lead to evasion of future vaccine induced protection, lessening the herd immunity potential of vaccination, similar to influenza A vaccines.
Mr. Otieno is a PhD student with the Virus Epidemiology and Control (VEC) Group (www.virec-group.org) at the Kenya Medical Research Institute (KEMRI)–Wellcome Trust Research Programme, Kilifi, Kenya. He is interested in the molecular evolutionary dynamics of viruses and how these inform on their transmission.
Acknowledgment
This work was supported by the Wellcome Trust, UK (grant nos. 102975, 100542, 084633, 077092). This article is published with permission from the director of the Kenya Medical Research Institute.
References
- Nair H, Nokes DJ, Gessner BD, Dherani M, Madhi SA, Singleton RJ, et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet. 2010;375:1545–55. DOIPubMedGoogle Scholar
- Nokes DJ, Okiro EA, Ngama M, Ochola R, White LJ, Scott PD, et al. Respiratory syncytial virus infection and disease in infants and young children observed from birth in Kilifi District, Kenya. Clin Infect Dis. 2008;46:50–7. DOIPubMedGoogle Scholar
- Agoti CN, Mwihuri AG, Sande CJ, Onyango CO, Medley GF, Cane PA, et al. Genetic relatedness of infecting and reinfecting respiratory syncytial virus strains identified in a birth cohort from rural Kenya. J Infect Dis. 2012;206:1532–41.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=22966119&dopt=Abstract DOIGoogle Scholar
- Cane PA, Pringle CR. Evolution of subgroup A respiratory syncytial virus: evidence for progressive accumulation of amino acid changes in the attachment protein. J Virol. 1995;69:2918–25.PubMedGoogle Scholar
- Mufson MA, Orvell C, Rafnar B, Norrby E. Two distinct subtypes of human respiratory syncytial virus. J Gen Virol. 1985;66:2111–24. DOIPubMedGoogle Scholar
- Peret TC, Hall CB, Schnabel KC, Golub JA, Anderson LJ. Circulation patterns of genetically distinct group A and B strains of human respiratory syncytial virus in a community. J Gen Virol. 1998;79:2221–9. DOIPubMedGoogle Scholar
- Agoti CN, Otieno JR, Ngama M, Mwihuri AG, Medley GF, Cane PA, et al. Successive respiratory syncytial virus epidemics in local populations arise from multiple variant introductions providing insights into virus persistence. J Virol. 2015;89:11630–42. DOIPubMedGoogle Scholar
- Otieno JR, Agoti CN, Gitahi CW, Bett A, Ngama M, Medley GF, et al. Molecular evolutionary dynamics of respiratory syncytial virus group A in recurrent epidemics in coastal Kenya. J Virol. 2016;90:4990–5002. DOIPubMedGoogle Scholar
- Cane PA. Molecular epidemiology of respiratory syncytial virus. Rev Med Virol. 2001;11:103–16. DOIPubMedGoogle Scholar
- Trento A, Galiano M, Videla C, Carballal G, García-Barreno B, Melero JA, et al. Major changes in the G protein of human respiratory syncytial virus isolates introduced by a duplication of 60 nucleotides. J Gen Virol. 2003;84:3115–20. DOIPubMedGoogle Scholar
- Trento A, Casas I, Calderón A, Garcia-Garcia ML, Calvo C, Perez-Breña P, et al. Ten years of global evolution of the human respiratory syncytial virus BA genotype with a 60-nucleotide duplication in the G protein gene. J Virol. 2010;84:7500–12. DOIPubMedGoogle Scholar
- Eshaghi A, Duvvuri VR, Lai R, Nadarajah JT, Li A, Patel SN, et al. Genetic variability of human respiratory syncytial virus A strains circulating in Ontario: a novel genotype with a 72 nucleotide G gene duplication. PLoS One. 2012;7:e32807. DOIPubMedGoogle Scholar
- Agoti CN, Otieno JR, Gitahi CW, Cane PA, Nokes DJ. Rapid spread and diversification of respiratory syncytial virus genotype ON1, Kenya. Emerg Infect Dis. 2014;20:950–9. DOIPubMedGoogle Scholar
- Pierangeli A, Trotta D, Scagnolari C, Ferreri ML, Nicolai A, Midulla F, et al. Rapid spread of the novel respiratory syncytial virus A ON1 genotype, central Italy, 2011 to 2013. Euro Surveill. 2014;19:20843. DOIPubMedGoogle Scholar
- Prifert C, Streng A, Krempl CD, Liese J, Weissbrich B. Novel respiratory syncytial virus a genotype, Germany, 2011-2012. Emerg Infect Dis. 2013;19:1029–30. DOIPubMedGoogle Scholar
- Auksornkitti V, Kamprasert N, Thongkomplew S, Suwannakarn K, Theamboonlers A, Samransamruajkij R, et al. Molecular characterization of human respiratory syncytial virus, 2010-2011: identification of genotype ON1 and a new subgroup B genotype in Thailand. Arch Virol. 2014;159:499–507. DOIPubMedGoogle Scholar
- Avadhanula V, Chemaly RF, Shah DP, Ghantoji SS, Azzi JM, Aideyan LO, et al. Infection with novel respiratory syncytial virus genotype Ontario (ON1) in adult hematopoietic cell transplant recipients, Texas, 2011-2013. J Infect Dis. 2015;211:582–9. DOIPubMedGoogle Scholar
- Valley-Omar Z, Muloiwa R, Hu N-C, Eley B, Hsiao N-Y. Novel respiratory syncytial virus subtype ON1 among children, Cape Town, South Africa, 2012. Emerg Infect Dis. 2013;19:668–70. DOIPubMedGoogle Scholar
- Tsukagoshi H, Yokoi H, Kobayashi M, Kushibuchi I, Okamoto-Nakagawa R, Yoshida A, et al. Genetic analysis of attachment glycoprotein (G) gene in new genotype ON1 of human respiratory syncytial virus detected in Japan. Microbiol Immunol. 2013;57:655–9.PubMedGoogle Scholar
- Duvvuri VR, Granados A, Rosenfeld P, Bahl J, Eshaghi A, Gubbay JB. Genetic diversity and evolutionary insights of respiratory syncytial virus A ON1 genotype: global and local transmission dynamics. Sci Rep. 2015;5:14268. DOIPubMedGoogle Scholar
- Hotard AL, Laikhter E, Brooks K, Hartert TV, Moore ML. Functional analysis of the 60 nucleotide duplication in the respiratory syncytial virus Buenos Aires strain attachment glycoprotein. J Virol. 2015;89:8258–66. DOIPubMedGoogle Scholar
- Nokes DJ, Ngama M, Bett A, Abwao J, Munywoki P, English M, et al. Incidence and severity of respiratory syncytial virus pneumonia in rural Kenyan children identified through hospital surveillance. Clin Infect Dis. 2009;49:1341–9. DOIPubMedGoogle Scholar
- Nokes DJ, Okiro EA, Ngama M, White LJ, Ochola R, Scott PD, et al. Respiratory syncytial virus epidemiology in a birth cohort from Kilifi district, Kenya: infection during the first year of life. J Infect Dis. 2004;190:1828–32. DOIPubMedGoogle Scholar
- Hammitt LL, Kazungu S, Welch S, Bett A, Onyango CO, Gunson RN, et al. Added value of an oropharyngeal swab in detection of viruses in children hospitalized with lower respiratory tract infection. J Clin Microbiol. 2011;49:2318–20. DOIPubMedGoogle Scholar
- Scott PD, Ochola R, Ngama M, Okiro EA, Nokes DJ, Medley GF, et al. Molecular epidemiology of respiratory syncytial virus in Kilifi district, Kenya. J Med Virol. 2004;74:344–54. DOIPubMedGoogle Scholar
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80. DOIPubMedGoogle Scholar
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30:2725–9. DOIPubMedGoogle Scholar
- Chernomor O, von Haeseler A, Minh BQ. Terrace Aware Data Structure for phylogenomic inference from supermatrices. Syst Biol. 2016;65:997–1008. DOIPubMedGoogle Scholar
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1. DOIPubMedGoogle Scholar
- Gupta R, Jung E, Brunak S. NetNGlyc: prediction of N-glycosylation sites in human proteins. 2004 [cited 12/23/16]. http://www.cbs.dtu.dk/services/NetNGlyc
- Kinyanjui TM, House TA, Kiti MC, Cane PA, Nokes DJ, Medley GF. Vaccine induced herd immunity for control of respiratory syncytial virus disease in a low-income country setting. PLoS One. 2015;10:e0138018. DOIPubMedGoogle Scholar
- Yoshihara K, Le MN, Okamoto M, Wadagni ACA, Nguyen HA, Toizumi M, et al. Association of RSV-A ON1 genotype with increased pediatric acute lower respiratory tract infection in Vietnam. Sci Rep. 2016;6:27856. DOIPubMedGoogle Scholar
- Panayiotou C, Richter J, Koliou M, Kalogirou N, Georgiou E, Christodoulou C. Epidemiology of respiratory syncytial virus in children in Cyprus during three consecutive winter seasons (2010-2013): age distribution, seasonality and association between prevalent genotypes and disease severity. Epidemiol Infect. 2014;142:2406–11. DOIPubMedGoogle Scholar
- Pretorius MA, van Niekerk S, Tempia S, Moyes J, Cohen C, Madhi SA, et al.; SARI Surveillance Group. Replacement and positive evolution of subtype A and B respiratory syncytial virus G-protein genotypes from 1997-2012 in South Africa. J Infect Dis. 2013;208(Suppl 3):S227–37. DOIPubMedGoogle Scholar
- Yu X, Kou Y, Xia D, Li J, Yang X, Zhou Y, et al. Human respiratory syncytial virus in children with lower respiratory tract infections or influenza-like illness and its co-infection characteristics with viruses and atypical bacteria in Hangzhou, China. J Clin Virol. 2015;69:1–6. DOIPubMedGoogle Scholar
- Kim YJ, Kim DW, Lee WJ, Yun MR, Lee HY, Lee HS, et al. Rapid replacement of human respiratory syncytial virus A with the ON1 genotype having 72 nucleotide duplication in G gene. Infect Genet Evol. 2014;26:103–12. DOIPubMedGoogle Scholar
- Khor CS, Sam IC, Hooi PS, Chan YF. Displacement of predominant respiratory syncytial virus genotypes in Malaysia between 1989 and 2011. Infect Genet Evol. 2013;14:357–60. DOIPubMedGoogle Scholar
- Balmaks R, Ribakova I, Gardovska D, Kazaks A. Molecular epidemiology of human respiratory syncytial virus over three consecutive seasons in Latvia. J Med Virol. 2014;86:1971–82. DOIPubMedGoogle Scholar
- Viegas M, Goya S, Mistchenko AS. Sixteen years of evolution of human respiratory syncytial virus subgroup A in Buenos Aires, Argentina: GA2 the prevalent genotype through the years. Infect Genet Evol. 2016;43:213–21. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 23, Number 2—February 2017
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
James R. Otieno, KEMRI–Wellcome Trust Research Programme, Kilifi, Kenya PO Box 230, 80108, Kilifi, Kenya
Top