Molecular Evolution, Diversity, and Adaptation of Influenza A(H7N9) Viruses in China
Jing Lu

, Jayna Raghwani
1, Rhys Pryce, Thomas A. Bowden, Julien Thézé, Shanqian Huang, Yingchao Song, Lirong Zou, Lijun Liang, Ru Bai, Yi Jing, Pingping Zhou, Min Kang, Lina Yi, Jie Wu
2, Oliver G. Pybus
2, and Changwen Ke
1
Author affiliations: Guangdong Provincial Center for Disease Control and Prevention, Guangzhou, China (J. Lu, Y. Song, L. Zou, L. Liang, R. Bai, Y. Jing, P. Zhou, M. Kang, L. Yi, J. Wu, C. Ke); Guangdong Provincial Institution of Public Health, Guangzhou (J. Lu, P. Zhou, L. Yi); University of Oxford, Oxford, UK (J. Raghwani, R. Pryce, T.A. Bowden, J. Thézé, O.G. Pybus); Beijing Normal University, Beijing, China (S. Huang)
Main Article
Figure 4
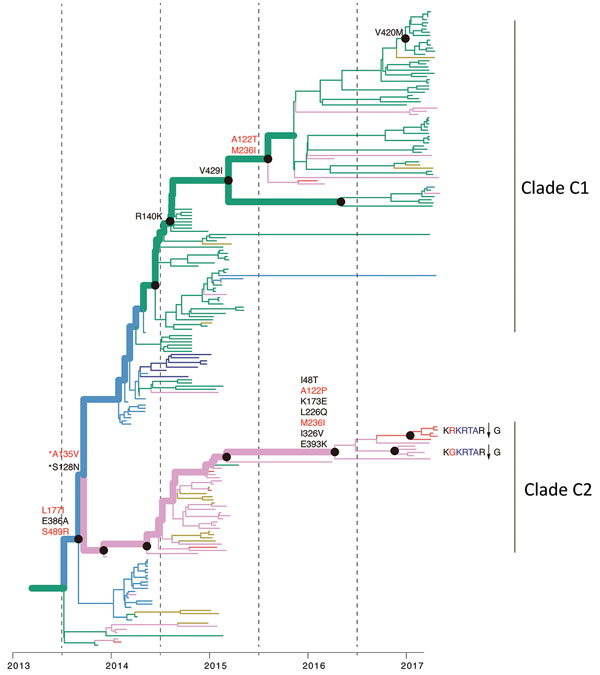
Figure 4. Reconstruction of amino acid changes along trunk of lineage B phylogenies of influenza A(H7N9) viruses, China. Maximum clade credibility tree of hemagglutinin gene sequences from lineage B is shown. Branches are colored according to geographic locations, as in Figure 3. Thicker lines indicate the trunk lineage leading up to the current fifth influenza epidemic wave. Amino acid changes along the trunk are indicated. Red branches indicate sites undergoing parallel amino acid changes across multiple lineages. Mutations correspond to H3 numbering scheme. *Indicates uncertainty about the phylogenetic position of the A135V and S128N mutations because branch posterior support is low.
Main Article
Page created: September 14, 2018
Page updated: September 14, 2018
Page reviewed: September 14, 2018
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.