Volume 24, Number 5—May 2018
Dispatch
Equine Encephalosis Virus in India, 2008
Cite This Article
Citation for Media
Abstract
A virus isolated from a sick horse from India in 2008 was confirmed by next-generation sequencing analysis to be equine encephalosis virus (EEV). EEV in India is concerning because several species of Culicoides midge, which play a major role in EEV natural maintenance and transmission, are present in this country.
Equine encephalosis is an arthropodborne, noncontagious disease of equids, characterized by fever and lassitude (1). For some affected horses, lack of appetite and some degree of edema have been reported as secondary complications (2). Equine encephalosis virus (EEV; family Reoviridae, genus Orbivirus) was first isolated from horses in South Africa in 1967 and was considered endemic to southern Africa until an outbreak was reported in Israel in 2008 (3). Reports of EEV circulation in Israel indicated that the virus is not limited to a particular region and raised concerns about the risk for its spread to other countries where competent hosts and vectors are present (3). A recent study examining EEV seroprevalence in Israel, Palestine, and Jordan has emphasized the potential risk for invasion of pathogens into new ecologic niches (4).
Two species of midge, Culicoides imicola (species complex) and C. bolitinos, have been implicated as EEV vectors (1). Although EEV infection results in high (60%–70%) morbidity rates among equids, deaths from this infection are rare (1). Diagnosis of EEV infection is confirmed by several techniques, including virus isolation in baby hamster kidney cells or suckling mice and demonstration of EEV antigen by antigen capture ELISA and serum virus neutralization tests. In addition, the TaqMan minor groove binder probe reverse transcription PCR has been used for rapid detection of EEV strains and differentiation from African horse sickness virus (5).
The EEV genome consists of 10 double-stranded RNA segments encapsulated by a double-layered icosahedral shell. The viral genome encodes 7 structural proteins (virus capsid proteins [VPs] 1–7) and 3 nonstructural proteins (NS1–3). On the basis of cross-neutralizing antibodies, 7 distinct serotypes of EEV (EEV-1–7) have been characterized (6).
Recent reports of next-generation sequencing analysis assert an unbiased identification of novel viruses in clinical samples (7,8). We investigated a virus isolated from necropsy samples received in 2008 from a horse in Pune, Maharashtra, India. Data analyzed from the MiniSeq (https://sapac.illumina.com/?langsel=/in/) output led to identification and generation of the complete EEV genome.
In July 2008, the National Institute of Virology in Pune received necropsy samples from a horse in Pune, including blood and organ tissue (lung, liver, kidney, and spleen). In addition, nasal swab, blood, and serum samples were collected from 13 horses on the same farm, reported to have clinical signs similar to those of the dead horse (fever, nasal discharge, loss of appetite, and weakness). These 13 horses recovered. No further specific details could be retrieved after this virus was identified.
All samples from the 14 horses were negative for equine influenza, Japanese encephalitis, and West Nile virus RNA according to PCR (9). We isolated virus by using horse tissue suspensions and blood samples as inocula. Each specimen was individually inoculated at a volume of 0.1 mL into 24-well plates containing a subconfluent monolayer of Vero CCL-81 cells. After incubating for 1 h at 37°C, the inoculum suspensions were removed and the cells rinsed twice with phosphate-buffered saline. We subsequently added Eagle minimum essential medium supplemented with 2% fetal calf serum and incubated the cells at 37°C. Postinfection cytopathic effects were observed by using an inverted light microscope (Nikon, Melville, NY, USA), and cellular morphologic changes were recorded. We determined viral titers in Vero CCL-81 cells to be 107.67 50% tissue culture infective dose/mL by using the method of Reed and Muench (10). We checked 8 vertebrate and invertebrate cell lines for susceptibility to this virus. Most cells, with the exception of bat embryonic cells and A549 (human lung carcinoma), displayed postinfection cytopathic effects (Table 1).
Because the initial identification attempts in 2008 yielded negative results, we performed a pathogen discovery protocol by using our recently acquired next-generation sequencing instrument. We performed DNA and RNA extractions separately on 1 mL of tissue culture supernatants or cell pellets of virus-infected cells by using a QIAamp DNA extraction kit or QIAamp Viral RNA extraction kit (QIAGEN, Valencia, CA, USA), according to manufacturer’s instructions. Concentrations of extracted RNA were quantified by using a Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA, USA), and host ribosomal RNA was depleted by using the NEBNext rRNA Depletion Kit (New England Biolabs, Ipswich, MA, USA). We requantified this purified RNA and prepared RNA libraries by using TruSeq Stranded mRNA Library Preparation Kit (Illumina, San Diego, CA, USA). Similarly, we prepared paired-end DNA sequencing libraries from DNA samples by using a Nextera XT DNA Library Prep Kit (Illumina). We quantified these libraries by using a KAPA Library Quantification Kit (Kapa Biosystems; Roche Diagnostics Corporation, Indianapolis, IN, USA) per manufacturer’s protocol and loaded them on an Illumina Miniseq next-generation sequencing platform. We imported the raw RNA data of 229 and 170 megabytes, along with DNA data of 176 and 199 megabytes for blood and lung samples, respectively, into the CLC Genomics Workbench software (QIAGEN) for analysis.
Figure 1
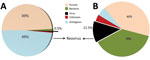
Figure 1. Sample identification by use of next-generation sequencing. RNA reads from blood (A) and lung (B) samples from horse that died of equine encephalosis in India, 2008. Samples were categorized by using...
To assemble contiguous sequences (contigs) by using de novo assembly, we used paired-end reads for DNA and RNA. DNA reads for blood and lung samples gave 166 and 113 contigs, with average lengths of 1,112 and 6,413 bp, respectively; 29 and 1,077 contigs were generated from the assembly of RNA reads for blood and lung samples, with average lengths of 1,376 and 1,034 bp, respectively. Figure 1 depicts data from the RNA reads, classified by using Taxonomer (11). BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) analysis of RNA contigs led to sample identification as EEV, and DNA contigs failed to identify any virus.
Figure 2

Figure 2. Phylogenetic tree of nonstructural 3 (A) and virus capsid protein 2 (B) genes of equine encephalosis virus. Kimura 2-parameter with (+ I) was used to create the evolutionary distance between 11...
After the infectious agent was identified, we conducted reference sequence mapping for the RNA reads by using the EEV isolate Potchefstroom with the help of a reference-guided assembly program (CLC Genomics Workbench). We recovered the full-genome sequence of EEV by using both contigs generated from de novo assembly and the reference mapping of the reads. A total of 7 fragments encoding the structural genes VP1–7 and 3 fragments encoding the genes NS1–3, together comprising ≈19,290 nt of EEV, were generated. We compared the obtained genome sequence with other publicly available EEV sequences (Table 2; Figure 2, panels A, B). NS3 gene analysis revealed that the EEV isolate from India shared the highest identity with the Potchefstroom EEV strain from South Africa, belonging to serotype 6. However, the VP2 gene had the highest resemblance to the Bryanston EEV strain, belonging to serotype 4.
The complete genome of the EEV isolated from a dead horse confirmed the presence of EEV in India. We compared the resultant full-genome sequence of this isolate with other EEV sequences available in GenBank. Phylogenetic analysis of the VP2 gene sequences, which is used for genotype classification of reoviruses, revealed that the isolate from India groups with the Bryanston EEV strain, belonging to serotype 4.
Because no specific treatments or vaccines for EEV are available, infected horses are given supportive treatment with nonsteroidal antiinflammatory drugs. The key control measure against EEV is to restrict its spread via arthropod vectors by carefully managing horses in a stable and adhering to appropriate biosafety measures. This report of EEV in India is concerning because several species of Culicoides midge, which play a major role in EEV natural maintenance and transmission, are present in this country.
This information about EEV has been provided to the Department of Animal Husbandry, Dairying, and Fisheries of the Ministry of Agriculture and Farmers Welfare in New Delhi, India. The Department of Animal Husbandry should soon initiate a survey to provide information about EEV presence in equine centers and commercial farms and to screen samples from sick horses. The concern is that this virus may be widely circulating in India without having been noticed earlier.
Dr. Yadav is the group leader for the Maximum Containment Laboratory, National Institute of Virology, Pune, India. Her research interests are novel pathogen discovery and emerging and reemerging zoonotic diseases of high-risk group viruses, including Crimean-Congo hemorrhagic fever, Kyasanur Forest disease, and Nipah.
Acknowledgments
We thank the authority of the horse stud farm in Pune for providing clinical samples. We also thank Rajen Lakra and Pravin Kore for their technical support during the study and Tatyana Klimova for editing this manuscript.
Financial support was provided by Centers for Disease Control and Prevention Global Health Security Agenda grant no. 6 NU2GGH001903-02-01 and intramural funding from the Indian Council of Medical Research, National Institute of Virology, Pune.
References
- Dhama K, Pawaiya RVS, Karthik K, Chakraborty S, Tiwari R, Verma AK. Equine encephalosis virus (EEV): a review. Asian J Anim Vet Adv. 2014;9:123–33. DOIGoogle Scholar
- McCall CA. Control of common infectious horse diseases [cited 2018 Feb 26]. http://www.aces.edu/pubs/docs/A/ANR-0589/index2.tmpl
- Westcott D, Mildenberg Z, Bellaiche M, McGowan SL, Grierson SS, Choudhury B, et al. Evidence for the circulation of equine encephalosis virus in Israel since 2001. PLoS One. 2013;8:e70532. DOIPubMedGoogle Scholar
- Tirosh-Levy S, Gelman B, Zivotofsky D, Quraan L, Khinich E, Nasereddin A, et al. Seroprevalence and risk factor analysis for exposure to equine encephalosis virus in Israel, Palestine and Jordan. Vet Med Sci. 2017;3:82–90. DOIPubMedGoogle Scholar
- Rathogwa NM, Quan M, Smit JQ, Lourens C, Guthrie AJ, van Vuuren M. Development of a real time polymerase chain reaction assay for equine encephalosis virus. J Virol Methods. 2014;195:205–10. DOIPubMedGoogle Scholar
- Aharonson-Raz K, Steinman A, Bumbarov V, Maan S, Maan NS, Nomikou K, et al. Isolation and phylogenetic grouping of equine encephalosis virus in Israel. Emerg Infect Dis. 2011;17:1883–6. DOIPubMedGoogle Scholar
- Yozwiak NL, Skewes-Cox P, Stenglein MD, Balmaseda A, Harris E, DeRisi JL. Virus identification in unknown tropical febrile illness cases using deep sequencing. PLoS Negl Trop Dis. 2012;6:e1485. DOIPubMedGoogle Scholar
- Prachayangprecha S, Schapendonk CM, Koopmans MP, Osterhaus AD, Schürch AC, Pas SD, et al. Exploring the potential of next-generation sequencing in detection of respiratory viruses. J Clin Microbiol. 2014;52:3722–30. DOIPubMedGoogle Scholar
- Shirato K, Miyoshi H, Kariwa H, Takashima I. Detection of West Nile virus and Japanese encephalitis virus using real-time PCR with a probe common to both viruses. J Virol Methods. 2005;126:119–25. DOIPubMedGoogle Scholar
- Reed LJ, Muench H. A simple method of estimating fifty per cent endpoints. Am J Hyg. 1938;27:493–7.
- Pyle RL. Taxonomer: a relational data model for managing information relevant to taxonomic research. PhyloInformatics. 2004;1:1–54.
Figures
Tables
Cite This ArticleTable of Contents – Volume 24, Number 5—May 2018
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Devendra T. Mourya, National Institute of Virology 20-A, Dr Ambedkar Rd, Pune, Maharashtra, Pin 411001, India; ,
Top