Volume 25, Number 11—November 2019
Dispatch
Isolation of Legionella pneumophila by Co-culture with Local Ameba, Canada
Cite This Article
Citation for Media
Abstract
Legionellosis was diagnosed in an immunocompromised 3-year-old girl in Canada. We traced the source of the bacterium through co-culture with an ameba collected from a hot tub in her home. We identified Legionella pneumophila serogroup 6, sequence type 185, and used whole-genome sequencing to confirm the environmental and clinical isolates were of common origin.
Legionella pneumophila is a waterborne bacterium responsible for Legionnaires’ disease, a potentially fatal respiratory disease acquired through environmental exposure to aerosolized water. According to the World Health Organization, L. pneumophila is the most common cause of legionellosis worldwide (1). For unknown reasons, cases reported in the United States and Europe have risen sharply over the past decade (2,3). Most legionellosis cases identified are caused by L. pneumophila serogroup 1 (4), possibly because of extensive use of initial urinary antibody screening that focuses on serogroup 1.
Free-living amebae are known natural environmental reservoirs for L. pneumophila (5). Amebae particularly play a role in legionellae growth in warm and stagnant engineered environments at 35°–45°C and in the bacterium’s persistence in high temperatures, biocides, and pH extremes (6). Co-culture with amebae is an efficient tool to detect L. pneumophila from human and environmental samples (7). However, amebae are rarely used in environmental investigations.
In December 2016, an immunocompromised 3-year-old girl was admitted to Alberta Children’s Hospital in Calgary, Alberta, Canada, with acute respiratory distress syndrome and septic shock requiring extracorporeal membrane oxygenation. She was transferred to a pediatric intensive care unit in Edmonton, Alberta, Canada, where examination of the lungs confirmed pneumonia in the left lower lobe segment with a lung abscess. Empiric treatment for infections was started immediately with meropenem, vancomycin, tobramycin, azithromycin, and trimethoprim/sulfamethoxazole. Subsequent bronchoalveolar lavage and bronchoscopy were performed, along with microbial culture for identification of fungi, mycobacteria, mycoplasma, viruses, and Legionella. Public Health Laboratory (ProvLab), Edmonton, successfully isolated Legionella sp. from clinical samples by using buffered charcoal yeast extract (BCYE) medium, with and without antimicrobial drugs, including polymyxin B, cycloheximide, and vancomycin. The clinical isolate was identified as L. pneumophila by using Legionella pneumophila Direct DFA Kit (Pro-Lab Diagnostics, https://pro-lab.com) direct fluorescent antibody assay. The National Microbiology Laboratory in Winnipeg, Manitoba, typed the isolate as serogroup 6, sequence type 185 (ST185), confirming Legionnaires’ disease.
The patient was treated with levofloxacin and a prophylactic dose of trimethoprim/sulfamethoxazole. She also received vancomycin, meropenem, and tobramycin for 8 days and azithromycin for 5 days. Her condition steadily improved, and she was discharged from the hospital a few days after treatment.
To investigate possible L. pneumophila sources, the Infection Prevention Control Research Laboratory of Alberta Health Services collected several first-flush water samples from sinks, a shower head, and a hot tub at the patient’s house and from sinks in the admitting hospital. All samples were negative for L. pneumophila by culture, but quantitative PCR results indicated the home hot tub was the likely source of the bacterium.
We initially attempted co-culture with Acanthamoeba polyphaga (ATCC30461) (8) but failed to isolate Legionella spp. Because growth of L. pneumophila in the environment is hypothesized to be dependent partly on the composition of local amebic populations (9), we isolated free-living amebae hosts from the hot tub samples. We then identified ameba-resisting bacteria (ARB) by using the isolated ameba in co-culture with the hot tub samples (7).
Figure 1
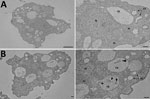
Figure 1. Transmission electron micrograph of amebae isolated from the home hot tub of a an immunocompromised 3-year-old girl with legionellosis before and after co-culture with Legionella pneumophila, Calgary, Alberta, Canada. A) Trophozoites...
We isolated free-living ameba hosts by filtering water from different environmental sites. We grew amebae at 30°C on nonnutrient agar supplemented with a thin film of viable Escherichia coli (ATCC25922). We isolated 2 free-living amebae, an Acanthamoeba sp. and a Vermamoeba vermiformis, and identified species by morphology (Figure 1, panel A) and 18S rRNA gene sequencing.
For co-culture experiments, we established amebae in axenic cultures in Nunc 25-cm2 tissue culture flasks (ThermoFisher Scientific, https://www.thermofisher.com) containing 5 mL serum casein glucose yeast extract medium at 37°C with 10% fetal calf serum. Before experiments, we performed subcultures of amebae every 3–4 days to ensure that trophozoites were in an exponential growth phase.
In brief, we co-cultured each environmental water sample with its isolated ameba by using several dilutions and incubating samples at 30°C for 12 h. When we observed amebal lysis, we recovered ARB on BCYE agar. We identified 1 of the ARB isolates from the ameba–hot tub culture as L. pneumophila by using 16S rRNA gene sequencing (Table) and subsequent sequence-based typing (10). Serotyping for L. pneumophila indicated both the clinical and environmental isolates were ST185, serogroup 6. We confirmed the presence of L. pneumophila inside V. vermiformis replicative phagosomes by transmission electron micrograph (Figure 1, panel B).
For confirmation, we performed whole-genome sequencing on clinical and environmental isolates. We extracted genomic DNA by using the NucleoSpin Tissue Kit (Macherey-Nagel, https://www.mn-net.com). We prepared libraries according to the protocol for the Nextera XT DNA Library Prep Kit (Illumina, https://www.illumina.com) and sequenced on an Illumina MiniSeq by using 2 × 150-nt reads. We deposited sequence information into BioProject (https://www.ncbi.nlm.nih.gov/bioproject) under accession no. PRJNA482644.
We trimmed sequence reads by using Trimmomatic version 0.36 (11) with the following parameters: Nextera clip, 2:30:10:8:true; LEADING, 20; TRAILING, 20; SLIDINGWINDOW, 4:20; MINLEN, 36. We assembled reads by using the SPAdes version 3.12 assembler in careful mode (12). We identified the closest related L. pneumophila strains for each set of contigs by using the PATRIC server (13) and searching for whole genome k-mer. We reordered assembled contigs by using mauve (14) against the closest matching reference, ATCC43290. We mapped trimmed sequence reads from both strains to the clinical isolate in the SPAdes de novo assembly by using Bowtie2 version 2.3.4.1 (https://github.com/BenLangmead/bowtie2), then processed reads with Picard (Broad Institute, https://www.broadinstitute.org) and identified single-nucleotide polymorphism insertions/deletions by using FreeBayes version 1.2.0 (https://www.geneious.com/plugins/freebayes). We filtered resulting polymorphisms with VCF.Filter version 4.2 (https://biomedical-sequencing.at/VCFFilter) by using QUAL >10 & DP >20 & QUAL/AO >10 & SAF >0 & SAR >0 & RPR >1 & RPL >1. To rule out assembly errors or other spurious calls, we visually inspected the location of each putative polymorphism in reference assemblies of each isolate and traced back to the clinical contigs.
We constructed a phylogenetic tree by submitting assembled scaffolds to the RAST server for genome annotation (15). To find core conserved orthologs with default parameters, we input the resulting gene annotations into OrthoMCL (https://orthomcl.org), alongside the complete ORFeomes of 6 other L. pneumophila strains: Philadelphia-1 NC_002942.5, Lens NC_006369.1, Thunder Bay CP003730.1, 570-CO-H NC_016811.1, Toronto-2005 NZ_CP012019.1, and Calgary-2012 SAMN03944918. We individually aligned 2,403 identified orthologs (2,471,034 nt) across all strains by using the MUSCLE algorithm (https://www.ebi.ac.uk/Tools/msa/muscle) and concatenated orthologs into a superalignment for tree construction. We adopted RAxML version 8.2.12 (ILRI Research Computing, http://hpc.ilri.cgiar.org) with a general time-reversible nucleotide substitution model for 1,000 bootstraps to generate a maximum-likelihood phylogenetic tree.
Figure 2
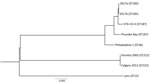
Figure 2. Phylogenetic tree depicting the relationship between Legionella pneumophila isolates identified during investigation of legionellosis in an immunocompromised 3-year-old girl, Calgary, Alberta, Canada, and reference sequences. L. pneumophila core ortholog-based maximum-likelihood phylogenetic...
Results of whole-genome sequencing analysis strongly suggest that clinical isolate 2017a and environmental isolate 2017b from the patient’s home hot tub were of common origin. With only a few single-nucleotide polymorphism differences (Figure 2), these data indicate the hot tub was the source of the patient’s infection.
Of note, we were not able to recover any legionellae from environmental samples with initial BCYE and other direct culture approaches. L. pneumophila serogroup 6 has not been identified in previous hot tub–associated infections, and this case might have been a result of the patient’s immunocompromised status. A commercial hot tub cleaning product was subsequently used for disinfection and maintenance of the hot tub. In a 1-year follow up analysis, water samples from the hot tub were negative for amebae and any Legionella spp.
Our report demonstrates the utility of ameba co-culture and emphasizes the use of locally sourced ameba to recover the source of L. pneumophila from environmental samples. Our findings also suggest that investigations should include free-living ameba to indicate the presence of potentially pathogenic Legionella spp. and as a potential factor to minimize the need for remediation actions associated with contaminated environments.
Dr. Dey is a researcher in the School of Public Health, University of Alberta, Edmonton, Canada. His research interests include free-living amebae and ameba-resisting bacteria.
Acknowledgments
The authors thank the Provincial Laboratory for Public Health (ProvLab), the Alberta Children’s Hospital in Calgary, and the Pediatric Intensive Care Unit in Edmonton. We also are grateful for the technical support and study suggestions provided by Lena Dlusskaya, Candis Scott, Shannon Braithwaite, Karen Hope, Kevin Fonseca, and Thomas Louie.
This study was supported by Alberta Innovates, Alberta, Canada, under grant no. 201300490.
Consent for publication: Written informed consent was obtained from the patient’s legal guardians for publication of this case report and any accompanying images.
Authors’ contributions: R.D. and N.J.A. conceived and designed the study; R.D. carried out the microbiome isolation and analysis; H.M. and A.W.E. sequenced the bacterium and analyzed the data; G.J.T. collected and analyzed the clinical data; L.P.W. collected the environmental samples; R.D. and N.J.A. wrote the manuscript with all authors contributing. All authors read and approved the final manuscript.
References
- World Health Organization. Legionellosis. 2018 Feb 16 [cited 2018 Feb 16]. https://www.who.int/en/news-room/fact-sheets/detail/legionellosis
- Beer KD, Gargano JW, Roberts VA, Reses HE, Hill VR, Garrison LE, et al. Outbreaks associated with environmental and undetermined water exposures—United States, 2011–2012. MMWR Morb Mortal Wkly Rep. 2015;64:849–51. DOIPubMedGoogle Scholar
- Beauté J; The European Legionnaires’ Disease Surveillance Network. Legionnaires’ disease in Europe, 2011 to 2015. Euro Surveill. 2017;22. DOIPubMedGoogle Scholar
- Cunha BA, Burillo A, Bouza E. Legionnaires’ disease. Lancet. 2016;387:376–85. DOIPubMedGoogle Scholar
- Rowbotham TJ. Current views on the relationships between amoebae, legionellae and man. Isr J Med Sci. 1986;22:678–89.
- Buse HY, Schoen ME, Ashbolt NJ. Legionellae in engineered systems and use of quantitative microbial risk assessment to predict exposure. Water Res. 2012;46:921–33. DOIPubMedGoogle Scholar
- Rowbotham TJ. Isolation of Legionella pneumophila serogroup 1 from human feces with use of amebic cocultures. Clin Infect Dis. 1998;26:502–3. DOIPubMedGoogle Scholar
- Pagnier I, Raoult D, La Scola B. Isolation and identification of amoeba-resisting bacteria from water in human environment by using an Acanthamoeba polyphaga co-culture procedure. Environ Microbiol. 2008;10:1135–44. DOIPubMedGoogle Scholar
- Dey R, Bodennec J, Mameri MO, Pernin P. Free-living freshwater amoebae differ in their susceptibility to the pathogenic bacterium Legionella pneumophila. FEMS Microbiol Lett. 2009;290:10–7. DOIPubMedGoogle Scholar
- Wong S, Pabbaraju K, Burk VF, Broukhanski GC, Fox J, Louie T, et al. Use of sequence-based typing for investigation of a case of nosocomial legionellosis. J Med Microbiol. 2006;55:1707–10. DOIPubMedGoogle Scholar
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. DOIPubMedGoogle Scholar
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77. DOIPubMedGoogle Scholar
- Wattam AR, Abraham D, Dalay O, Disz TL, Driscoll T, Gabbard JL, et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 2014;42(D1):D581–91. DOIPubMedGoogle Scholar
- Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5:
e11147 . DOIPubMedGoogle Scholar - Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. DOIPubMedGoogle Scholar
Figures
Table
Cite This ArticleOriginal Publication Date: October 03, 2019
Table of Contents – Volume 25, Number 11—November 2019
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Rafik Dey, University of Alberta School of Public Health; Rm 3-57D, South Academic Bldg; Edmonton, AB T6G 2G7, Canada
Top