Phenotypic and Genomic Analyses of Burkholderia stabilis Clinical Contamination, Switzerland
Helena M.B. Seth-Smith

, Carlo Casanova, Rami Sommerstein, Dominik M. Meinel
1, Mohamed M.H. Abdelbary
2, Dominique S. Blanc, Sara Droz, Urs Führer, Reto Lienhard, Claudia Lang, Olivier Dubuis, Matthias Schlegel, Andreas Widmer, Peter M. Keller
3, Jonas Marschall, and Adrian Egli
Author affiliations: University Hospital Basel, Basel, Switzerland (H.M.B. Seth-Smith, D.M. Meinel, A. Widmer, A. Egli); University of Basel, Basel (H.M.B. Seth-Smith, D.M. Meinel, A. Egli); University of Bern, Bern, Switzerland (C. Casanova, S. Droz); Bern University Hospital, Bern (R. Sommerstein, J. Marschall); Lausanne University Hospital, Lausanne, Switzerland (M.M.H. Abdelbary, D.S. Blanc); Biel Hospital, Biel, Switzerland (U. Führer); ADMED, La Chaux-de-Fonds, Switzerland (R. Lienhard); Viollier AG, Allschwil, Switzerland (C. Lang, O. Dubuis); Cantonal Hospital St. Gallen, St. Gallen, Switzerland (M. Schlegel); Swissnoso, National Center for Infection Prevention, Bern (M. Schlegel, A. Widmer, J. Marschall); University of Zurich, Zurich, Switzerland (P.M. Keller)
Main Article
Figure 2
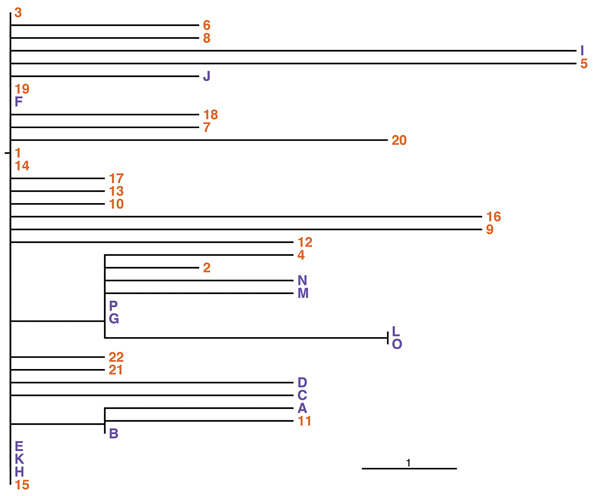
Figure 2. Phylogeny of outbreak isolates of Burkholderia stabilis strain CH16 from Switzerland based on high-quality single nucleotide polymorphisms (SNPs). This phylogeny of all sequenced outbreak isolates might represent a conservative estimate of SNP numbers. Given the large genome size and possible mismapping to repeats, it is difficult to determine the ultimate number of SNPs between samples. This phylogeny was confirmed using several parameters and manual checking of called SNPs. The root was arbitrarily chosen to give the fewest root to tip SNPs (n = 6). Numbers represent isolates from patients; letters represent isolates from washing gloves, located in the root position. Scale bar indicates 1 SNP.
Main Article
Page created: May 20, 2019
Page updated: May 20, 2019
Page reviewed: May 20, 2019
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.