Volume 25, Number 6—June 2019
Dispatch
Multirecombinant Enterovirus A71 Subgenogroup C1 Isolates Associated with Neurologic Disease, France, 2016–2017
Cite This Article
Citation for Media
Abstract
In 2016, an upsurge of neurologic disease associated with infection with multirecombinant enterovirus A71 subgenogroup C1 lineage viruses was reported in France. These viruses emerged in the 2000s; 1 recombinant is widespread. This virus lineage has the potential to be associated with a long-term risk for severe disease among children.
Figure 1
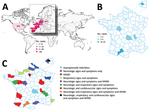
Figure 1. Geographic locations and numbers of enterovirus A71 (EV-A71) subgenogroup C1v2015 infections reported during 2015–2017. A) Countries in which EV-A71 C1v2015 was reported. The year the virus was first reported is indicated....
Enterovirus A71 (EV-A71) comprises 7 genogroups (A–G) and various subgenogroups (e.g., B0–B5, C1–C5) (1). B4, B5, and C4 viruses circulate mainly in Asia, and C1 and C2 viruses have been detected in Europe (2). In 2016, an upsurge in neurologic manifestations of enterovirus infection was reported in France (3). These cases were associated with an emerging lineage of subgenogroup C1 enteroviruses first reported in 2015 in Germany and later in Spain and 4 other countries (Figure 1, panel A) (4–8). Our aim was to obtain the full genomes of the viruses from the specimens collected in France and track down the origin of this emerging lineage, hereafter referred to as C1v2015.
According to consolidated data recorded from the French Enterovirus Surveillance Network, 77 laboratory-confirmed cases of C1v2015 infection occurred during March–October 2016; in comparison, 136 EV-A71 infections of all genogroups combined were recorded during 2010–2015. The C1v2015 cases were widespread throughout France and associated with various clinical manifestations, including meningitis, cerebellitis, encephalitis, and myelitis, as well as hand, foot and mouth disease (HFMD) (Figure 1, panels B, C). One fatal case resulted from HFMD and cardiorespiratory failure. We analyzed 32 clinical specimens available from 25 patients reported as having a C1v2015 infection in 2016 and 2017 (Table 1; Appendix Table 1). Specimens and clinical data were collected during routine clinical work-up and epidemiologic surveillance, and patient data were deidentified before this study was conducted. The study was approved by the review board Comité de Protection des Personnes Sud-Est VI (no. 2018/CE44) in Clermont-Ferrand, France. The study population comprised 16 hospitalized children (median age 0.1 years), 4 children seen via ambulatory care (median age 1.8 years), and 5 children with asymptomatic infection (median age 1.4 years) in a childcare facility placed under community surveillance. We obtained the complete genomes, including the full 5′ and 3′ untranslated regions (UTRs), of 18 of 20 specimens and partial genomes of 2 of 20 specimens (2,893-nt and 4,380-nt long) acquired from 18 children (Appendix) (2). We also determined the genomes of 12 isolates recovered during routine enterovirus surveillance to investigate their genetic relationships with C1v2015 (Appendix Table 2); we selected these viruses on the basis of previous exploratory investigations of their partial sequences (2,9,10).
Figure 2

Figure 2. Nucleotide similarity and phylogenetic analyses of EV-A71 subgenogroup C1v2015 isolates, France, 2016–2017, constructed to determine temporal origin of C1v2015 lineage. A) Nucleotide similarity patterns between EV-A71 C1v2015 and other EV-A lineages...
We performed whole-genome sequence analyses as previously described (11) to identify which viruses were the closest relatives of C1v2015. The C1v2015 genome appears to be a mosaic comprising 4 modules defined by distinct patterns of similarity possibly arising through recombination (Figure 2, panel A). The nucleotide similarity patterns for module 2 (genomic region P1 comprising 4 capsid protein genes) suggest this region was inherited en bloc from an earlier subgenogroup C1 lineage. We used genomic region P1 to determine the evolutionary relatedness between C1v2015 and earlier C1 viruses and to date when the upsurge of C1v2015 infections began in Europe (Figure 2, panel B). All C1v2015 viruses clustered in a lineage distinct from that comprising the C1 viruses reported during 1991–2010. The nucleotide substitution rate of C1v2015 (5.2238 [95% highest probability density HPD interval 4.124–6.3737] × 10–3 nt substitutions/y) and earlier C1 lineages (4.6302 [95% HPD interval 4.1769–5.1353] × 10–3 nt substitutions/y) was similar. All of the P1 sequences from these viruses, except that of the virus from patient 14, had a maximum nucleotide sequence difference from each other of 2%; the P1 sequence of the virus from patient 14 differed from that of other C1v2015 viruses by 4.8%. The close genetic relatedness between the C1v2015 sequences reported during 2015–2017 in France, Germany, Japan, and the United States was indicative of rapid widespread transmission. We estimated that interpersonal transmission of this lineage began during 2009–2011 (Table 2; Figure 2, panel B) and that its spread was sustained during 2013–2014, just 1–2 years before C1v2015 was first reported. The most recent common ancestor between C1v2015 and earlier C1 viruses was dated to 2000–2002. Seven EV-A71 subgenogroup C1 viruses from Africa and Europe were located at the base of the C1v2015 lineage (Figure 2, panel B), suggesting that the C1 strain involved in the emergence of C1v2015 was circulating in this region during the 2000s.
The C1v2015 genomic module 4 comprises the entire 3Dpol gene and has a 90%–95% nucleotide similarity with 4 distinct EV-A genomes: coxsackievirus A2 (CV-A2) and CV-A5 from Russia, CV-A4 from China, and CV-A6 from Turkmenistan (Figure 2, panel A). We performed another phylogenetic analysis to assess the temporal origin of C1v2015 using this module. With the 3Dpol phylogenetic analysis, we estimated that C1v2015 began spreading in 2010–2014 (Table 2; Figure 2, panel C), an estimate similar to that calculated with the P1 phylogeny. The nucleotide substitution rates with this analysis were also similar (C1v2015 3.7689 [95% HPD interval 1.3003–6.5838] × 10–3 nt substitutions/y and C1 3.6318 [95% HPD interval 1.6064–6.2072] × 10–3 nt substitutions/y). Whole-genome sequencing analysis showed that the isolate from patient 14 (14|COC286037|FRA|2016) shared distinct 3Dpol genes with other C1v2015 viruses (Appendix Figure 1). Overall, data indicate that the virus from patient 14 was an early recombinant of the C1v2015 lineage (Appendix Figure 2).
Within genomic module 1 (5′ UTR, first 600 nt), we found areas of moderate nucleotide similarity (90%–95%) between the C1v2015 genome and the CV-A6 and CV-A8 genomes and lower similarity (<88%) with the EV-A71 subgenogroup C1 genomes (Figure 2, panel A). The C1v2015 5′ UTR was therefore inherited from an EV-A lineage virus but not from the C1 ancestors that provided the capsid region. The pattern of sequence variation in the 5′ UTR precludes the possibility of analysis with a molecular clock.
The genomic module 3 of C1v2015 had low similarity with all the publicly available EV-A genomes; thus, the precise origin remains unknown (Figure 2, panel A). The highest nucleotide similarity scores (<90% with CV-A5 genomes) indicate only a distant genetic relationship. We conclude that genes 2A (except the 5′ terminus), 2B, 2C, and 3A–3C were transferred into the C1v2015 genome from a previously unreported lineage.
Thirty years after the outbreaks in central Europe (12,13), the 2016 upsurge of infections is a reminder that EV-A71 is of growing public health concern. After the B5 and C4 subgenogroup upsurges, C1v2015 is the latest example of an emerging recombinant EV-A71 associated with neurologic manifestations. Recombination, which frequently occurs in enteroviruses, is considered a factor driving this viral emergence (14,15). Compared with earlier circulating lineages of EV-A71, C1v2015 is a multirecombinant that arose through complete shuffling of all nonstructural genomic regions, although the capsid genes are phylogenetically typical of C1 viruses. Shuffling involved >2 recombination events with EV-A genomes before the emergence of C1v2015 as a life-threatening pathogen (Appendix Figure 2). From a public health perspective, the spread of C1v2015 could have resulted from acquired genomic features, notably a unique combination of the 5′ UTR and 3Dpol gene, because recombination events clearly preceded the extensive circulation of C1v2015. The mosaic structure of the genome indicates that C1v2015 is an integral part of a large recombination network including multiple EV-A viruses transmitted in Eurasia. Given the propensity of enteroviruses to recombine their genomes and spread rapidly across distant countries (2,11) and that C1v2015 circulation continued throughout 2017 and 2018 in France, we need to determine if this virus is associated with a long-term recurrent risk for severe disease in the pediatric population through sharing data from global surveillance.
Ms. Tomba Ngangas is a doctoral candidate at the Université Clermont Auvergne in Clermont-Ferrand, France. Her research interests are in characterizing viruses that cause neurologic disease and hand, foot and mouth disease.
Acknowledgments
The authors are indebted to Patrice Bouissou, Annick Givois, and Martine Wagner-Vaucard, who obtained samples in children with hand, foot and mouth disease. We acknowledge the technical contribution of Jeroen Cremer, Adeline Duard, Nathalie Rodde, and Isabelle Simon for helpful assistance with molecular typing and sequencing. We thank Jeffrey Watts for help in preparing the manuscript in English.
Financial support was provided by the University Clermont Auvergne, France.
References
- Bessaud M, Razafindratsimandresy R, Nougairède A, Joffret ML, Deshpande JM, Dubot-Pérès A, et al. Molecular comparison and evolutionary analyses of VP1 nucleotide sequences of new African human enterovirus 71 isolates reveal a wide genetic diversity. PLoS One. 2014;9:
e90624 . DOIPubMedGoogle Scholar - Hassel C, Mirand A, Lukashev A, TerletskaiaLadwig E, Farkas A, Schuffenecker I, et al. Transmission patterns of human enterovirus 71 to, from and among European countries, 2003 to 2013. Euro Surveill. 2015;20:30005. DOIPubMedGoogle Scholar
- Antona D, Kossorotoff M, Schuffenecker I, Mirand A, Leruez-Ville M, Bassi C, et al. Severe paediatric conditions linked with EV-A71 and EV-D68, France, May to October 2016. Euro Surveill. 2016;21:30402. DOIPubMedGoogle Scholar
- Böttcher S, Obermeier PE, Neubauer K, Diedrich S; Laboratory Network for Enterovirus Diagnostics. Recombinant enterovirus A71 subgenogroup C1 strains, Germany, 2015. Emerg Infect Dis. 2016;22:1843–6. DOIPubMedGoogle Scholar
- Karrasch M, Fischer E, Scholten M, Sauerbrei A, Henke A, Renz DM, et al. A severe pediatric infection with a novel enterovirus A71 strain, Thuringia, Germany. J Clin Virol. 2016;84:90–5. DOIPubMedGoogle Scholar
- Casas-Alba D, de Sevilla MF, Valero-Rello A, Fortuny C, García-García JJ, Ortez C, et al. Outbreak of brainstem encephalitis associated with enterovirus-A71 in Catalonia, Spain (2016): a clinical observational study in a children’s reference centre in Catalonia. Clin Microbiol Infect. 2017;23:874–81. DOIPubMedGoogle Scholar
- Wieczorek M, Purzyńska M, Krzysztoszek A, Ciąćka A, Figas A, Szenborn L. Genetic characterization of enterovirus A71 isolates from severe neurological cases in Poland. J Med Virol. 2018;90:372–6. DOIPubMedGoogle Scholar
- Midgley SE, Nielsen AG, Trebbien R, Poulsen MW, Andersen PH, Fischer TK. Co-circulation of multiple subtypes of enterovirus A71 (EV- A71) genotype C, including novel recombinants characterised by use of whole genome sequencing (WGS), Denmark 2016. Euro Surveill. 2017;22:30565. DOIPubMedGoogle Scholar
- Lukashev AN, Shumilina EY, Belalov IS, Ivanova OE, Eremeeva TP, Reznik VI, et al. Recombination strategies and evolutionary dynamics of the Human enterovirus A global gene pool. J Gen Virol. 2014;95:868–73. DOIPubMedGoogle Scholar
- van der Sanden S, van der Avoort H, Lemey P, Uslu G, Koopmans M. Evolutionary trajectory of the VP1 gene of human enterovirus 71 genogroup B and C viruses. J Gen Virol. 2010;91:1949–58. DOIPubMedGoogle Scholar
- Hassel C, Mirand A, Farkas A, Diedrich S, Huemer HP, Peigue-Lafeuille H, et al.; HFMD French Study Network. Phylogeography of coxsackievirus A16 reveals global transmission pathways and recent emergence and spread of a recombinant genogroup. J Virol. 2017;91:
e00630-17 . DOIPubMedGoogle Scholar - Chumakov M, Voroshilova M, Shindarov L, Lavrova I, Gracheva L, Koroleva G, et al. Enterovirus 71 isolated from cases of epidemic poliomyelitis-like disease in Bulgaria. Arch Virol. 1979;60:329–40. DOIPubMedGoogle Scholar
- Nagy G, Takátsy S, Kukán E, Mihály I, Dömök I. Virological diagnosis of enterovirus type 71 infections: experiences gained during an epidemic of acute CNS diseases in Hungary in 1978. Arch Virol. 1982;71:217–27. DOIPubMedGoogle Scholar
- McWilliam Leitch EC, Cabrerizo M, Cardosa J, Harvala H, Ivanova OE, Koike S, et al. The association of recombination events in the founding and emergence of subgenogroup evolutionary lineages of human enterovirus 71. J Virol. 2012;86:2676–85. DOIPubMedGoogle Scholar
- Kyriakopoulou Z, Pliaka V, Amoutzias GD, Markoulatos P. Recombination among human non-polio enteroviruses: implications for epidemiology and evolution. Virus Genes. 2015;50:177–88. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleOriginal Publication Date: April 30, 2019
Table of Contents – Volume 25, Number 6—June 2019
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Jean-Luc Bailly, Université Clermont Auvergne, LMGE UMR 6023, Clermont-Ferrand, France
Top