Volume 27, Number 11—November 2021
Research
Multidrug-Resistant Methicillin-Resistant Staphylococcus aureus Associated with Bacteremia and Monocyte Evasion, Rio de Janeiro, Brazil
Cite This Article
Citation for Media
Abstract
We typed 600 methicillin-resistant Staphylococcus aureus (MRSA) isolates collected in 51 hospitals in the Rio de Janeiro, Brazil, metropolitan area during 2014–2017. We found that multiple new clonal complex (CC) 5 sequence types had replaced previously dominant MRSA lineages in hospitals. Whole-genome analysis of 208 isolates revealed an emerging sublineage of multidrug-resistant MRSA, sequence type 105, staphylococcal cassette chromosome mec II, spa t002, which we designated the Rio de Janeiro (RdJ) clone. Using molecular clock analysis, we hypothesized that this lineage began to expand in the Rio de Janeiro metropolitan area in 2009. Multivariate analysis supported an association between bloodstream infections and the CC5 lineage that includes the RdJ clone. Compared with other closely related isolates, representative isolates of the RdJ clone more effectively evaded immune function related to monocytic cells, as evidenced by decreased phagocytosis rate and increased numbers of viable unphagocytosed (free) bacteria after in vitro exposure to monocytes.
Methicillin-resistant Staphylococcus aureus (MRSA) is characterized by the mainly clonal structure of bacterial populations and the worldwide spread of a few highly successful lineages, sequence types (STs), and clonal complexes (CCs) that cycle through waves of dominance (1,2). During the late 1990s, the Brazilian endemic clone (BEC), which belongs to the ST239(CC8)–staphylococcal cassette chromosome (SCC) mecIII lineage, comprised ≈80% of MRSA isolates in hospitals in Brazil (3). In the 2000s, isolates of the ST1(CC1)-SCCmecIV lineage supplanted BEC in >2 hospitals in the Rio de Janeiro metropolitan area of Brazil (4). More recent analyses have suggested that CC5 isolates might be increasing in prevalence in Brazil (5).
Most studies on the molecular epidemiology of MRSA in Brazil have analyzed a small number of isolates from a limited number of hospitals (5–9). We used molecular and genomic approaches to characterize 600 MRSA isolates collected from 51 hospitals in the Rio de Janeiro metropolitan area and identified a novel MRSA clone of ST105-SCCmecII spa t002 (ST105-SCCmecII-t002), which we termed the Rio de Janeiro (RdJ) clone, as a predominant cause of MRSA bloodstream infections (BSIs).
Bacterial Isolates
We obtained the MRSA isolates from 600 patients at 51 hospitals in the Rio de Janeiro metropolitan area and confirmed MRSA using routine identification methods (Table 1; Appendix 1). The sample comprised roughly equal numbers of isolates from blood samples from BSI patients, nonblood samples from patients with infections at another body site, and nasal swab samples; samples were collected during 2014–2017, most in 2015 and 2016. Patient age was available for 450 patients (Table 2). The research protocols were submitted to the Human Research Ethics Committee (CAAE submission no. 41614914.4.00005257) of the Hospital Universitário Clementino Fraga Filho, Universidade Federal do Rio de Janeiro (Rio de Janeiro, Brazil); the study was considered non–human subject research.
Molecular Typing and Susceptibility Testing
We used restriction-modification (RM) tests to determine CC (10) and multiplex PCR to type SCCmec (11). We used PCR to screen for the lukSF-PV, agrII, SCCmecIII, and seh genes as previously described (12). We conducted antibiogram and susceptibility tests for glycopeptide drugs as recommended by Clinical and Laboratory Standards Institute guidelines (13).
Genome Sequencing and Analysis
We selected 208 isolates for whole-genome sequencing (WGS). Because of a strong predominance (179/208; 86.1%) of CC5 isolates, we focused our research on the CC5 lineage. We randomly selected isolates from blood (70/145; 48.3%), nonblood (52/114; 45.6%), and nasal swab (57/123; 46.3%) CC5 samples (Appendix 2). The other 29 isolates used in WGS belonged to less abundant CCs. We prepared genomic DNA using the Wizard Genomic DNA Purification Kit (Promega Corporation, https://www.promega.com) and sequenced genome libraries by using Nextera XT DNA Library Prep Kit (Illumina, https://www.illumina.com) and the HiSeq 2500 system (Illumina) using paired-end reads of 125 bp. We trimmed reads using BBDuk Trimmer version 1.0 (Geneious, https://www.geneious.com) and assembled genomes using Velvet Assembly version 7.0.4 (14) and SPAdes version 3.13.0 (15). We used RAST (https://rast.nmpdr.org) and manual inspection to annotate the isolates. We determined the genotypes of the sequenced strains using the MLST 2.0, SCCmecFinder 1.2, and spa Typer 1.0 tools (https://cge.cbs.dtu.dk).
Phylogenetic Analysis and Divergence Times
We constructed a maximum-likelihood tree for 661 CC5 genomes: 179 genomes from the current investigation and 482 assembled genomes available on GenBank, chosen from the list provided by Challagundla et al. (8) (Appendix 3). We used a single-nucleotide polymorphism (SNP) alignment produced by Snippy to infer an initial phylogenetic tree in RAxML version 8.2.4 (16).
To estimate when the ST105-SCCmecII-t002 lineage emerged in Rio de Janeiro, we used a Bayesian phylogenetic framework to analyze 73 genomes that passed our Mash Screen (17) quality cutoffs. We selected MRSA strain FCFHV36, the closest complete reference genome available in GenBank, using the WhatsGNU topgenome (-t) option (18). We used the SNP alignment to infer an initial phylogenetic tree in RAxML version 8.2.4 before using ClonalFrameML (19) to detect and mask areas of recombination. We used the SNP recombination-masked alignment to estimate divergence times in BEAST version 2.6.2 (20). We found a positive correlation between genetic divergence and isolation time using TempEst version 1.5.3 (21). We plotted the chronograms based on the maximum clade credibility tree using the TreeAnnotator program and visualized in FigTree version 1.4.3 (Appendix 3).
Genomic Island Characterization
Figure 1
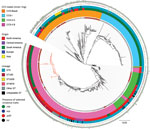
We used Geneious Prime version 2020.1.2 to manually inspect the ΦSA3, vSa-α, vSa-β, vSa-ɣ, and SaPI-1 genomic islands (22,23) and Swiss-Prot (Uniprot Consortium, https://www.uniprot.org) to annotate paralogues. To map the genetic context of genomic islands, we randomly selected representative genome sequences from different phylogenetic locations of the tree showing the most common CC5 lineages in the Rio de Janeiro metropolitan area (Figure 1). We determined gene presence or absence using BLAST analysis (https://blast.ncbi.nlm.nih.gov).
Phagocytosis Assays
We subjected the selected isolates to phagocytosis (Appendix 3 Table 1). In this assay, we considered the entire process of phagocytosis (i.e., binding and uptake) by detecting all cell-associated bacteria, whether internalized or externally attached, after washing. We cultured bacteria at 37°C for 18 h at 250 rpm in brain–heart infusion broth (Becton Dickinson, https://www.bd.com) before treating with 25 nmol SYTO 9 stain (Thermo Fisher Scientific, https://www.thermofisher.com) for 15 min and washing in phosphate-buffered saline (1× phosphate-buffered saline, pH 7.2). We incubated bacterial cells at 37°C for 30 min in 5% carbon dioxide with THP-1 monocytes in Roswell Park Memorial Institute 1640 medium for a multiplicity of infection of 10 (24). We did not use antimicrobial drugs at any time during these assays. We washed the infected monocytes with PBS once and then centrifuged them at 200 × g for 5 min. We resuspended THP-1 cells in PBS and analyzed them by flow cytometry (FACSCalibur; Becton Dickinson). We acquired 10,000 live THP-1 cells (as calculated by forward scatter and side scatter gating) and analyzed data using FlowJo10 software (https://www.flowjo.com). We calculated the number of bacteria-associated THP-1 cells as the frequency of fluorescent (i.e., SYTO 9–positive) THP-1 cells compared with total live THP-1 cells. In addition, we counted and compared the number of viable unphagocytosed bacterial cells in the culture supernatant of each assay at 0 and 30 min after incubation.
Statistical Analyses
We analyzed molecular typing, antimicrobial testing, and epidemiologic data using Pearson χ2 tests. To assess the association of the CC5-SCCmecII group and the ST105-SCCmecII-t002 sublineage with BSI, we used Stata 16.0 (https://www.stata.com) to conduct a Mantel-Haenszel test stratified on a composite variable informed by participant age (>60 years vs. <60 years), year of specimen collection (2014, 2015, or 2016–2017), and hospital type (public vs. private). For the analysis of year of specimen collection, we combined data from 2016 and 2017 because few isolates were collected during 2017. We analyzed phagocytosis assays using a 1-way analysis of variance and Tukey multiple comparison test in GraphPad Prism 6 (GraphPad Software, Inc., https://www.graphpad.com).
Distribution of Genotypes (CC-SCCmec) and Antimicrobial Resistance
Figure 2
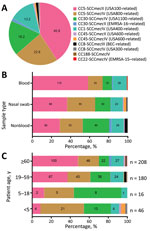
Figure 2. Distribution of 600 MRSA isolates by lineage (A), sample type (B), and patient age (C), Rio de Janeiro Brazil, 2014–2017. A) MRSA isolates by lineage (CC-SCCmectype) among 600...
Among the 600 isolates that underwent CC and SCCmec typing, most were categorized as CC5-SCCmecII (245/600; 40.8%) or CC5-SCCmecIV (137/600; 22.8%). The second most common lineage was CC30, comprised of lukSF-PV–positive CC30-SCCmecIV (109/600; 18.2%) and lukSF-PV–negative CC30-SCCmecII (8/600; 1.3%) isolates. The previously dominant CC1-SCCmecIV lineage (79/600; 13.2%) and BEC clone (7/600; 1.2%) were much less frequent. In addition, we observed low frequencies of STs related to other international lineages such as CC45-SCCmecII/IV (related to USA600), CC8-SCCmecIV (related to USA300), and CC22-SCCmecIV (related to EMRSA-15) (Figure 2, panel A).
Compared with isolates of other frequent clonal lineages, CC5-SCCmecII isolates were more likely to be multidrug-resistant, defined as having resistance to >4 non-β-lactam antimicrobial drugs (48.6% vs. 5.8%) (Table 3). In contrast, CC5-SCCmecIV strains showed more susceptibility to non–β-lactams; only 4.4% were multidrug-resistant. All 109 strains belonging to the CC30-SCCmecIV lineage, which is related to the community-acquired MRSA USA1100/Oceania South West Pacific clone, were susceptible to all non–β-lactams tested (Table 3).
Distribution of Genotypes (CC-SCCmec) and Clinical Data
In the univariate analysis, we found that the distribution of genotypes was associated with MRSA infection site (Figure 2, panel B). CC5-SCCmecII isolates were more common among blood (115/245; 46.9%) than nonblood (62/245; 25.3%) and nasal swab (68/245; 27.8%) samples, whereas CC5-SCCmecIV isolates were more common among nasal swab (55/137; 40.1%) and nonblood (52/137; 38.0%) than blood (30/137; 21.9%) samples. The third most frequent lineage, CC30-SCCmecIV, was more common among nonblood (60/109; 55.0%) than nasal swab (30/109; 27.5%) and blood (19/109; 17.4%) samples.
The distribution of MRSA lineages varied among age groups. CC5-SCCmecII was more common among patients >60 years of age (100/208; 48.1%). CC30-SCCmecIV prevalence was higher among younger populations and diminished with increasing age range; prevalence was 50.0% (8/16) among children 5–18 years of age, 21.1% (38/180) among adults 19–59 years of age, and 10.6% (22/208) among adults >60 years of age. Among children <5 years of age, the most prevalent lineage was CC5-SCCmecIV, which is sometimes known as the pediatric clone (21/46; 45.7%) (Figure 2, panel C). Adults 19–59 and >60 years of age had a similar prevalence of CC5-SCCmecIV isolates (23.9% among adults 19–59 years of age vs. 22.1% among adults >60 years of age). The proportion of CC5-SCCmecII isolates was also similar between adults 19–59 years of age (57/180; 31.7%) and adults >60 years of age (62/208; 29.8%). CC5-SCCmecII was associated with BSIs even after stratifying for the composite variable of age, hospital type, and year of isolation (p<0.01).
Novel MRSA Clone
To better characterize the circulating clones, especially those belonging to CC5, we used whole-genome sequencing on 208 isolates: 76 (36.5%) from blood samples, 69 (33.2%) from nasal swab samples, and 63 (30.3%) from nonblood samples. Most (179; 86.1%) isolates belonged to CC5, whereas 29 did not (Appendix 3 Table 2). Multilocus and spa-typing using WGS revealed 4 CC5 clones that constituted >75% of isolates (Table 4). The dominant genotype, ST105(CC5)-SCCmecII-t002, the RdJ clone, comprised 41.9% (75/179) of the CC5 isolates. RdJ showed the second highest proportion of multidrug resistance (41/75; 54.7%), superseded only by ST5-SCCmecII-t539 (14/17; 82.4%). In contrast, only 1 (2.3%) strain of ST5-SCCmecIV-t002 was multidrug-resistant. MRSA lineages coexisting in the same hospital often displayed different resistance profiles.
In addition to being the most frequent MRSA clone, RdJ might be responsible for the higher frequency of CC5-SCCmecII isolates from blood samples. ST105-SCCmecII-t002 isolates were more common among blood (41/75; 54.7%) than nasal swab (20/75; 26.7%) and nonblood (14/75; 18.7%) samples; however, when adjusted for hospital type and year of isolation, this association became nonsignificant (p = 0.12).
Whole-Genome Phylogenetic Analysis of MRSA CC5 Isolates
Figure 3
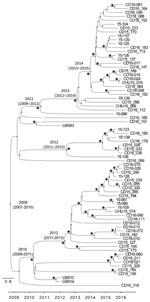
Figure 3. Time-calibrated phylogenetic tree of methicillin-resistant Staphylococcus aureus ST105(CC5)-SCCmecII-t002 lineage, Rio de Janeiro, Brazil, 2014–2017. Chronogram constructed using Bayesian phylogenetic analysis of single-nucleotide polymorphisms from 73 genomes....
The whole-genome phylogenetic analysis grouped CC5 isolates from this study into 3 of the 4 major phylogenetic groups corresponding mostly to the ST105(CC5)-SCCmecII-t002, ST5-SCCmecII-t539, and ST5-SCCmecIV-t002 genotypes and distributed widely throughout the CC5 tree (Figure 1). All SCCmecIV isolates clustered in the CC5-Basal clade. Isolates with the multidrug-resistant ST5(CC5)-SCCmecII-t539 genotype clustered with members of the paraphyletic group CC5-IIA described by Challagundla et al. (8). Most other CC5 isolates, including isolates of genotype ST105(CC5)-SCCmecII-t002, were grouped in clade CC5-IIB. Although most of these isolates form the RdJ clade, which is found mostly in Rio de Janeiro, nearby outgroups to this clade are composed of previously sequenced isolates from São Paulo and Porto Alegre (25) and North America, as well as a few isolates from this study (Appendix 3). This pattern might indicate multiple introductions into Brazil. Our Bayesian analysis of the RdJ clade suggests a recent date of introduction, probably 2009 (95% highest posterior density 2007–2010) (Figure 3).
In comparison with other CC5 genomes, the clade that includes the ST105 genomes lacked key virulence genes. In addition to the apparent loss of the enterotoxin P gene (sep) noted by Challagundla et al. (8), isolates from this clade uniformly lacked the splD gene encoding serine protease D (Figure 1).
Monocytic Evasion
Figure 4
Figure 4. Scatter plot representing gating strategy for identifying monocytes in the FSC-H versus SSC-H analysis of methicillin-resistant Staphylococcus aureusisolates, Rio de Janeiro, Brazil, 2014–2017. Representative flow cytometry chart shows the...
Figure 5

Figure 5. Histograms showing count versus green fluorescence intensity of THP-1 cells exposed or not to MRSA isolates, Rio de Janeiro, Brazil, 2014–2017. A) Acquisition of THP-1 cells not exposed to MRSA. B)...
Figure 6

Figure 6. Scatter plots showing evasion of phagocytosis by MRSA isolates, Rio de Janeiro, Brazil, 2014–2017. A) Phagocytosis rates for representatives of the phylogenetic groups CC5-Basal, CC5-IIA, and CC5-IIB. Four independent experiments...
To better ascertain differences in pathogenicity of RdJ isolates, we assessed the in vitro phagocytosis rate and viable counts of unphagocytosed (free) bacteria (Figures 4,5,6). Representative RdJ isolates showed very low rates of phagocytosis/host cell association (2.9%) compared with representatives of other CC5 lineages: 41.3% for ST5(CC5)-SCCmecII-t539 and 35.8% for ST5(CC5)-SCCmecIV-t002 strains (Figure 6, panel A). In addition, after a 30-minute interaction with THP-1 monocytes, the RdJ strains showed higher survival rates (5.58%) than other lineages: 0.88% for ST5(CC5)-SCCmecIV-t002 and 0.76% for ST5(CC5)-SCCmecII-t539 (Figure 6, panel B).
Using molecular typing and phylogenetic analysis, we identified a third epidemic lineage of MRSA in Rio de Janeiro. CC5, and to a lesser extent CC30, have become the most prevalent MRSA lineages in Rio de Janeiro hospitals, replacing the previously dominant ST1(CC1)-SCCmecIV lineage, which had replaced the BEC lineage ST239(CC8)-SCCmecIII during 2004–2008 (4). At the time when the ST1(CC1)-SCCmecIV lineage replaced BEC, CC5 comprised only 10% of isolates (4); CC5 now constitutes >60% of isolates. Previously dominant clones, especially BEC, carried resistance to many non–β-lactam antimicrobial drugs, antiseptics, and heavy metals whereas the currently dominant strains are more susceptible (1).
Although CC5-SCCmecII was the predominant genotype in our sample, the proportions of the second and third most frequent genotypes, CC5-SCCmecIV and lukSF-PV–positive CC30-SCCmecIV, also had increased from prior studies (4). CC5-SCCmecIV (related to USA800), which was first isolated in children at a hospital in Portugal in 1992 (26), was overrepresented among patients <5 years of age in our sample. Some studies have suggested that this strain is more common among children (27), although the nature of this association remains unclear. The lukSF-PV–positive CC30-SCCmecIV genotype is related to the USA1100/Oceania South West Pacific clone (1), and is a distant relative of the historically epidemic and especially virulent phage type 80/81 lineage (28). We previously showed that, in contrast to the 80/81 lineage, ST30(CC30)-SCCmecIV MRSA from Brazil displays a natural attenuation of the Agr and SaeRS virulence regulators (29), which might explain why this lineage was responsible for only 9.6% of BSIs in this study.
The large number of MRSA isolates genotyped in this study enabled us to assess the distribution of MRSA genotypes by patient age and sites of infection or colonization. We identified associations between the CC5-SCCmecII genotype, BSIs, and older age, possibly because of the increased virulence or invasiveness of this genotype. The CC5-SCCmecII genotype also is found in the USA100 lineage ST5(CC5)-SCCmecII that was dominant among hospitals in the United States during the late 1990s (30), before the emergence of the USA300 clone (31). USA100 is still found in hospitals in the United States (32) and around the world (1).
In our sample, most (75/114; 65.8%) CC5-SCCmecII isolates belonged to ST105 and shared spa-type t002, suggesting the emergence of a new clone. ST105(CC5)-SCCmecII strains have previously infected humans and domestic animals (33), and 4 isolates from this lineage were reported in a hospital in São Paulo (7). Reports from other countries have occasionally shown a substantial prevalence of this lineage, including a study that showed colonization among 22.4% of patients admitted to a hospital in Pennsylvania, USA (34). Another study showed that ST105(CC5)-SCCmecII was the predominant lineage among patients who had MRSA BSI in Switzerland (35). In Portugal, ST105(CC5)-SCCmecII has been reported as the most abundant MRSA colonizing patients >60 years of age (33); a multicenter study identified this lineage as the second most common clone among patients who had BSIs (36). The first vancomycin-resistant S. aureus isolate in Portugal belonged to this lineage (37), a troubling finding because most vancomycin-resistant S. aureus isolates have belonged to the CC5 lineage (25).
Few studies exist on the molecular epidemiology of MRSA in Brazil and in other countries from South America; existing studies are based on a limited number of samples (5–7,9). As a result, the full extent of the dissemination of the ST105-SCCmecII-t002 genotype in Latin America is unknown. Since the late 2000s, ST105-SCCmecII-t002 has been reported as the second or third most frequent MRSA lineage in hospitals in the United States and some countries in Europe (33–35). For example, researchers documented an outbreak of ST105-SCCmecII-t002 MRSA among 18 neonates at Mount Sinai Hospital (New York, NY, USA) during 2014–15 (38). In addition, ST105 isolates comprised 87.5% of delafloxacin-resistant MRSA strains collected in 7 hospitals in New York (39). Altogether, these data show that ST105 is a major MRSA lineage not only in Rio de Janeiro but also in other countries. ST105-SCCmecII-t002 also might have spread in other regions of Brazil; therefore, more studies are needed to better track and investigate this lineage.
We used Bayesian molecular clock analysis to estimate the expansion of the RdJ clade in Rio de Janeiro in 2009 (95% highest posterior density 2007–2010), which is consistent with previous estimates that date the origin of the ST105 lineage to the mid-1990s (8). The ST105 clade is characterized by a lack of virulence genes that are common among other CC5 strains. All ST105(CC5)-SCCmecII isolates lacked the sep gene encoding enterotoxin P, as noted by Challagundla et al. (8). In addition to its emetic properties, enterotoxin P is a superantigen that induces T-cell proliferation and production of proinflammatory cytokines (40). ST105(CC5)-SCCmecII-t002 strains showed resistance to fluoroquinolones, macrolides, and lincosamides. ST105(CC5)-SCCmecII isolates also lacked the serine protease encoding gene splD, despite the presence of the splABCF genes of the spl operon. Although the specific role of SplD in S. aureus pathogenesis is not known, some researchers have proposed that Spl serine proteases might use proteolysis to modulate host proteins critical to bacterial pathogenesis (41). Future work should address implications of the absence of SplD in the ST105(CC5)-SCCmecII lineage.
Compared with representatives of the ST5-SCCmecII-t539 and ST5-SCCmecIV-t002 lineages, representative isolates of the RdJ clade showed increased evasion of phagocytosis mechanisms upon exposure to monocytic cells (i.e., THP-1). Multiple factors, including phagocytosis rate and the activity of toxic compounds released by monocytes, might affect the number of viable unphagocytosed bacterial cells (42). Moreover, we observed an increased number of viable RdJ free cells. The basis of this phenotype is unclear and deserves further study. Le Pabic et al. (43) implicated the small noncoding RNA, SprC, and its effect on regulation of the major autolysin Atl in S. aureus evasion of phagocytosis by human monocytes and macrophages. However, we did not find any differences in the sprC gene of the 6 representative strains tested, suggesting that the observed evasion might be multifactorial, probably linked to the production of several bacterial molecules (42).
One limitation of this study is the lack of more extensive clinical data such as the presence of indwelling catheters or lines and underlying conditions that might have affected our estimates. The association between ST105-SCCmecII-t002 and BSIs was attenuated when accounting for other variables such as hospital type and year of isolation, but still might be of clinical relevance. Access to more extensive clinical data would enable further exploration of this relationship. In addition, our reliance on samples from Rio de Janeiro might have affected our phylogenetic analysis; focused sampling in other geographic locations might show a more widespread epidemic.
In summary, we uncovered a new MRSA clone in hospitals in the Rio de Janeiro metropolitan area. Our findings emphasize the dynamic nature of the local rise and decline of various MRSA clones. In addition, these data indicate that MRSA clonal dynamics also might be associated with different manifestations of disease and host factors, such as age. This analysis revealed the emergence of a novel multidrug-resistant MRSA clone associated with BSIs. This association might be critical for assessing the clinical and epidemiologic risks associated with the spread of this clone and the biologic basis for its putative enhanced invasiveness.
Ms. Viana is a doctoral candidate at Federal University of Rio de Janeiro, Rio de Janeiro, Brazil. Her main research interests are genomics and phylogenetic evolution of methicillin-resistant Staphylococcus aureus.
Acknowledgments
We thank the microbiology department of Diagnósticos da América S.A. for providing the bacterial isolates included in this work.
This work was supported in part by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq grant no. 140774/2019-0 to A.S.V. and grant nos. 443804/2018-4 and 307672/2019-0 to A.M.S.F.), Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (grant no. E-26/210.193/2014 to A.M.S.F. and P.J.P.; grant nos. E-26/010.001280/2016, E-26.202.803/2017, and E-26/010.002435/2019 to A.M.S.F.); the Bill & Melinda Gates Foundation (grant no. OPP1193112 to A.M.S.F.) and the US National Institutes of Health (grant no. 1R01AI137526-01 to P.J.P. and A.M.M.).
References
- Figueiredo AMS, Ferreira FA. The multifaceted resources and microevolution of the successful human and animal pathogen methicillin-resistant Staphylococcus aureus. Mem Inst Oswaldo Cruz. 2014;109:265–78. DOIPubMedGoogle Scholar
- Planet PJ, Narechania A, Chen L, Mathema B, Boundy S, Archer G, et al. Architecture of a species: phylogenomics of Staphylococcus aureus. Trends Microbiol. 2017;25:153–66. DOIPubMedGoogle Scholar
- Teixeira LA, Resende CA, Ormonde LR, Rosenbaum R, Figueiredo AM, de Lencastre H, et al. Geographic spread of epidemic multiresistant Staphylococcus aureus clone in Brazil. J Clin Microbiol. 1995;33:2400–4. DOIPubMedGoogle Scholar
- Silva-Carvalho MC, Bonelli RR, Souza RR, Moreira S, dos Santos LCG, de Souza Conceição M, et al. Emergence of multiresistant variants of the community-acquired methicillin-resistant Staphylococcus aureus lineage ST1-SCCmecIV in 2 hospitals in Rio de Janeiro, Brazil. Diagn Microbiol Infect Dis. 2009;65:300–5. DOIPubMedGoogle Scholar
- Chamon RC, Ribeiro SD, da Costa TM, Nouér SA, Dos Santos KRN. Complete substitution of the Brazilian endemic clone by other methicillin-resistant Staphylococcus aureus lineages in two public hospitals in Rio de Janeiro, Brazil. Braz J Infect Dis. 2017;21:185–9. DOIPubMedGoogle Scholar
- Zuma AVP, Lima DF, Assef APDC, Marques EA, Leão RS. Molecular characterization of methicillin-resistant Staphylococcus aureus isolated from blood in Rio de Janeiro displaying susceptibility profiles to non-β-lactam antibiotics. Braz J Microbiol. 2017;48:237–41. DOIPubMedGoogle Scholar
- Caiaffa-Filho HH, Trindade PA, Gabriela da Cunha P, Alencar CS, Prado GVB, Rossi F, et al. Methicillin-resistant Staphylococcus aureus carrying SCCmec type II was more frequent than the Brazilian endemic clone as a cause of nosocomial bacteremia. Diagn Microbiol Infect Dis. 2013;76:518–20. DOIPubMedGoogle Scholar
- Challagundla L, Reyes J, Rafiqullah I, Sordelli DO, Echaniz-Aviles G, Velazquez-Meza ME, et al. Phylogenomic classification and the evolution of clonal complex 5 methicillin-resistant Staphylococcus aureus in the Western Hemisphere. Front Microbiol. 2018;9:1901. DOIPubMedGoogle Scholar
- Arias CA, Reyes J, Carvajal LP, Rincon S, Diaz L, Panesso D, et al. A prospective cohort multicenter study of molecular epidemiology and phylogenomics of Staphylococcus aureus bacteremia in nine Latin American countries [Erratum in: Antimicrob Agents Chemother. 2017;61:e00095–18]. Antimicrob Agents Chemother. 2017;61:17. DOIGoogle Scholar
- Cockfield JD, Pathak S, Edgeworth JD, Lindsay JA. Rapid determination of hospital-acquired meticillin-resistant Staphylococcus aureus lineages. J Med Microbiol. 2007;56:614–9. DOIPubMedGoogle Scholar
- Milheiriço C, Oliveira DC, de Lencastre H. Update to the multiplex PCR strategy for assignment of mec element types in Staphylococcus aureus [Erratum in: Antimicrob Agents Chemother. 2007;51:4537]. Antimicrob Agents Chemother. 2007;51:3374–7.DOIPubMedGoogle Scholar
- Beltrame CO, Botelho AMN, Silva-Carvalho MC, Souza RR, Bonelli RR, Ramundo MS, et al. Restriction modification (RM) tests associated to additional molecular markers for screening prevalent MRSA clones in Brazil. Eur J Clin Microbiol Infect Dis. 2012;31:2011–6. DOIPubMedGoogle Scholar
- Clinical and Laboratory Standards Institute. Performance standards for antimicrobial susceptibility testing: 28th informational supplement (M100-S28). Wayne (PA): The Institute; 2018.
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–9. DOIPubMedGoogle Scholar
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77. DOIPubMedGoogle Scholar
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3. DOIPubMedGoogle Scholar
- Ondov BD, Starrett GJ, Sappington A, Kostic A, Koren S, Buck CB, et al. Mash Screen: high-throughput sequence containment estimation for genome discovery. Genome Biol. 2019;20:232. DOIPubMedGoogle Scholar
- Moustafa AM, Planet PJ. WhatsGNU: a tool for identifying proteomic novelty. Genome Biol. 2020;21:58. DOIPubMedGoogle Scholar
- Didelot X, Wilson DJ. ClonalFrameML: efficient inference of recombination in whole bacterial genomes. PLOS Comput Biol. 2015;11:
e1004041 . DOIPubMedGoogle Scholar - Bouckaert R, Vaughan TG, Barido-Sottani J, Duchêne S, Fourment M, Gavryushkina A, et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLOS Comput Biol. 2019;15:
e1006650 . DOIPubMedGoogle Scholar - Rambaut A, Lam TT, Max Carvalho L, Pybus OG. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016;2:
vew007 . DOIPubMedGoogle Scholar - Aswani V, Najar F, Pantrangi M, Mau B, Schwan WR, Shukla SK. Virulence factor landscape of a Staphylococcus aureus sequence type 45 strain, MCRF184. BMC Genomics. 2019;20:123. DOIPubMedGoogle Scholar
- Novick RP, Ram G. Staphylococcal pathogenicity islands-movers and shakers in the genomic firmament. Curr Opin Microbiol. 2017;38:197–204. DOIPubMedGoogle Scholar
- Melehani JH, James DBA, DuMont AL, Torres VJ, Duncan JA. Staphylococcus aureus leukocidin A/B (LukAB) kills human monocytes via host NLRP3 and ASC when extracellular, but not intracellular. PLoS Pathog. 2015;11:
e1004970 . DOIPubMedGoogle Scholar - Panesso D, Planet PJ, Diaz L, Hugonnet JE, Tran TT, Narechania A, et al. Methicillin-susceptible, vancomycin-resistant Staphylococcus aureus, Brazil. Emerg Infect Dis. 2015;21:1844–8. DOIPubMedGoogle Scholar
- Sá-Leão R, Santos Sanches I, Dias D, Peres I, Barros RM, de Lencastre H. Detection of an archaic clone of Staphylococcus aureus with low-level resistance to methicillin in a pediatric hospital in Portugal and in international samples: relics of a formerly widely disseminated strain? J Clin Microbiol. 1999;37:1913–20. DOIPubMedGoogle Scholar
- Rokney A, Baum M, Ben-Shimol S, Sagi O, Anuka E, Agmon V, et al. Dissemination of the methicillin-resistant Staphylococcus aureus pediatric clone (ST5-T002-IV-PVL+) as a major cause of community-associated staphylococcal infections in Bedouin children, southern Israel. Pediatr Infect Dis J. 2019;38:230–5. DOIPubMedGoogle Scholar
- DeLeo FR, Kennedy AD, Chen L, Bubeck Wardenburg J, Kobayashi SD, Mathema B, et al. Molecular differentiation of historic phage-type 80/81 and contemporary epidemic Staphylococcus aureus. Proc Natl Acad Sci U S A. 2011;108:18091–6. DOIPubMedGoogle Scholar
- Ramundo MS, Beltrame CO, Botelho AMN, Coelho LR, Silva-Carvalho MC, Ferreira-Carvalho BT, et al. A unique SaeS allele overrides cell-density dependent expression of saeR and lukSF-PV in the ST30-SCCmecIV lineage of CA-MRSA. Int J Med Microbiol. 2016;306:367–80. DOIPubMedGoogle Scholar
- Roberts RB, Chung M, de Lencastre H, Hargrave J, Tomasz A, Nicolau DP, et al.; Tri-State MRSA Collaborative Study Group. Distribution of methicillin-resistant Staphylococcus aureus clones among health care facilities in Connecticut, New Jersey, and Pennsylvania. Microb Drug Resist. 2000;6:245–51. DOIPubMedGoogle Scholar
- Planet PJ. Life after USA300: the rise and fall of a superbug. J Infect Dis. 2017;215(suppl_1):S71–7. DOIPubMedGoogle Scholar
- Carrel M, Perencevich EN, David MZ. USA300 methicillin-resistant Staphylococcus aureus, United States, 2000–2013. Emerg Infect Dis. 2015;21:1973–80. DOIPubMedGoogle Scholar
- Almeida ST, Nunes S, Paulo ACS, Faria NA, de Lencastre H, Sá-Leão R. Prevalence, risk factors, and epidemiology of methicillin-resistant Staphylococcus aureus carried by adults over 60 years of age. Eur J Clin Microbiol Infect Dis. 2015;34:593–600. DOIPubMedGoogle Scholar
- Verghese B, Schwalm ND III, Dudley EG, Knabel SJ. A combined multi-virulence-locus sequence typing and Staphylococcal Cassette Chromosome mec typing scheme possesses enhanced discriminatory power for genotyping MRSA. Infect Genet Evol. 2012;12:1816–21. DOIPubMedGoogle Scholar
- Blanc DS, Petignat C, Wenger A, Kuhn G, Vallet Y, Fracheboud D, et al. Changing molecular epidemiology of methicillin-resistant Staphylococcus aureus in a small geographic area over an eight-year period. J Clin Microbiol. 2007;45:3729–36. DOIPubMedGoogle Scholar
- Faria NA, Miragaia M, de Lencastre H; Multi Laboratory Project Collaborators. Massive dissemination of methicillin resistant Staphylococcus aureus in bloodstream infections in a high MRSA prevalence country: establishment and diversification of EMRSA-15. Microb Drug Resist. 2013;19:483–90. DOIPubMedGoogle Scholar
- Melo-Cristino J, Resina C, Manuel V, Lito L, Ramirez M. First case of infection with vancomycin-resistant Staphylococcus aureus in Europe. Lancet. 2013;382:205. DOIPubMedGoogle Scholar
- Sullivan MJ, Altman DR, Chacko KI, Ciferri B, Webster E, Pak TR, et al. A complete genome screening program of clinical methicillin-resistant Staphylococcus aureus isolates identifies the origin and progression of a neonatal intensive care unit outbreak. J Clin Microbiol. 2019;57:e01261–19. DOIPubMedGoogle Scholar
- Iregui A, Khan Z, Malik S, Landman D, Quale J. Emergence of delafloxacin-resistant Staphylococcus aureus in Brooklyn, New York. Clin Infect Dis. 2020;70:1758–60. DOIPubMedGoogle Scholar
- Omoe K, Imanishi K, Hu D-L, Kato H, Fugane Y, Abe Y, et al. Characterization of novel staphylococcal enterotoxin-like toxin type P. Infect Immun. 2005;73:5540–6. DOIPubMedGoogle Scholar
- Zdzalik M, Kalinska M, Wysocka M, Stec-Niemczyk J, Cichon P, Stach N, et al. Biochemical and structural characterization of SplD protease from Staphylococcus aureus. PLoS One. 2013;8:
e76812 . DOIPubMedGoogle Scholar - Flannagan RS, Heit B, Heinrichs DE. Antimicrobial mechanisms of macrophages and the immune evasion strategies of Staphylococcus aureus. Pathogens. 2015;4:826–68. DOIPubMedGoogle Scholar
- Le Pabic H, Germain-Amiot N, Bordeau V, Felden B. A bacterial regulatory RNA attenuates virulence, spread and human host cell phagocytosis. Nucleic Acids Res. 2015;43:9232–48. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleOriginal Publication Date: October 01, 2021
1These first authors contributed equally to this article.
2Current affiliation: Fluminense Federal University, Niteroi, Brazil.
3Current affiliation: Fundação Oswaldo Cruz, Rio de Janeiro, Brazil.
4These authors were co–principal investigators.
Table of Contents – Volume 27, Number 11—November 2021
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Paul J. Planet, Division of Pediatric Infectious Diseases, Children’s Hospital of Philadelphia, Philadelphia, PA 19104, USA
Top