Volume 28, Number 2—February 2022
Research
Role of Anopheles Mosquitoes in Cache Valley Virus Lineage Displacement, New York, USA
Cite This Article
Citation for Media
Abstract
Cache Valley virus (CVV) is a mosquitoborne virus that infects livestock and humans. We report results of surveillance for CVV in New York, USA, during 2000–2016; full-genome analysis of selected CVV isolates from sheep, horse, humans, and mosquitoes from New York and Canada; and phenotypic characterization of selected strains. We calculated infection rates by using the maximum-likelihood estimation method by year, region, month, and mosquito species. The highest maximum-likelihood estimations were for Anopheles spp. mosquitoes. Our phylogenetic analysis identified 2 lineages and found evidence of segment reassortment. Furthermore, our data suggest displacement of CVV lineage 1 by lineage 2 in New York and Canada. Finally, we showed increased vector competence of An. quadrimaculatus mosquitoes for lineage 2 strains of CVV compared with lineage 1 strains.
Cache Valley virus (CVV; family Peribunyaviridae, genus Orthobunyavirus) belongs to the order Bunyavirales, which consists of 12 families and 46 genera that are major human, livestock, and plant pathogens (1). CVV contains negative-sense, single-stranded RNA organized into 3 separate segments designated large (L), medium (M), and small (S) (2). The L RNA segment encodes the RNA-dependent RNA polymerase (L protein); the M segment encodes 2 glycoproteins, Gn and Gc, which are inserted in the viral membrane, plus a nonstructural protein; and the S segment encodes the nucleocapsid protein and a second nonstructural protein (3).
CVV isolates fall into 2 lineages. Lineage 1 viruses were isolated in the United States and Canada during 1956–2011, and lineage 2 consists of more recent strains from the northeastern United States (4). Lineage 2 was shown to have displaced lineage 1 in Connecticut, USA, during 2010–2014 (4). CVV is widespread throughout North and Central America and infects sheep, cattle, white-tailed deer, and humans (4). The virus has been isolated from >30 mosquito species in several genera; however, the principal vectors remain unknown (5). Accumulating evidence from surveillance suggests that Anopheles quadrimaculatus and An. punctipennis mosquitoes might be major vectors of CVV (6).
We performed surveillance of CVV during 2000‒2016 in New York. We also determined vector competence of An. quadrimaculatus for representative CVV strains.
Mosquito Collection
Figure 1
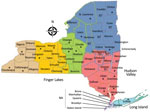
Figure 1. Counties in New York, USA, in which Cache Valley virus was studied during 2000‒2016 (https://www.health.ny.gov/statistics/cancer/registry/images/nycounty). Asterisks (*) indicate counties in which samples positive for Cache Valley virus were...
Mosquitoes were submitted from the following regions in New York: West, Finger Lakes, North, Central, Hudson Valley, and Long Island (Figure 1). Mosquitoes were collected by local health districts by using Centers for Disease Control and Prevention light traps with CO2 (7) or gravid (8) traps. We identified mosquitoes to species morphologically (9), and pooled females into groups of ≈50 mosquitoes by trap type, date collected, and trap location. Mosquitoes were transported on dry ice to the Arbovirus Laboratories, Wadsworth Center, New York State Department of Health (Slingerlands, NY, USA), for testing and were stored at −80°C until processed.
Virus Isolation
We processed mosquito pools as described (10,11). In brief, we homogenized pools in 1 mL of mosquito diluent containing 20% fetal bovine serum, 50 μg of streptomycin/mL, 50 U of penicillin, and 2.5 μg of amphotericin B/mL in phosphate-buffered saline in a Retsch Mixer Mill (https://www.retsch.com) set to 24 cycles/s for 2 min. We used viral stocks of 2 CVV strains isolated from cerebrospinal fluid of humans (strain Hu-2022) and from brain tissue (strain PA) (12,13) for RNA extraction. We used RNA extracted from brain tissues of a horse that died from neurologic disease and tested positive for CVV in this analysis. We homogenized placenta tissues from sheep (from a ewe that had given birth to a deformed lamb in a southern Ontario flock during 2011) and used them to infect Vero E6 cells for virus isolation. Cytopathic effect was observed after 6 days, and supernatant was harvested and used for RNA extraction and to generate virus stocks.
RNA Extraction
We used extraction plates (Thermofisher, https://www.thermofisher.com), which were prepared on a Tecan Evo 150 Liquid Handler (Tecan, https://www.tecan.com) and used 50 μL of homogenates or viral stocks to extract RNA on a Magmax 96 Express (Applied Biosystems, https://www.thermofisher.com) and a MagMax Viral Isolation Kit (Thermofisher). A total of 90 μL of homogenized sample RNA was eluted.
Primer Design and Reverse Transcription PCR
We used a standard PCR to identify CVV isolates as described (14). Beginning in 2012, we developed a real-time reverse transcription PCR (RT-PCR) by using new primers and probes (CVVF1, CVVR1, and CVV1 probe) to expedite the surveillance process (Table 1). A quantitative RT-PCR was developed according to manufacturer’s protocol (Quanta Biosciences, https://www.quantabio.com) with slight modifications. The final volume of the reaction was 15 μL and consisted of 10 μL of master mixture and 5 μL of template. Each reaction contained 0.7 μmol/L of each forward and reverse primers and 0.3 μmol/L of probe. We performed real-time quantitation by using ABI Prism 7500 (Life Technologies, https://www.thermofisher.com). Cycling conditions were as follows: 3 min at 50°C, followed by 10 min at 95°C, then 40 cycles of alternating 95°C for 10 s and 60°C for 30 s. After introduction of CVV lineage 2, we developed new primers and probes (CVVF2, CVVR2, and CVV2 probe) for better detection (Table 1).
Maximum-Likelihood Estimation
We used maximum-likelihood estimation calculations to determine prevalence of CVV in mosquitoes. These calculations were based on a program developed by Brad Biggerstaff (https://www.cdc.gov/westnile/resourcepages/mosqsurvsoft.html).
Sequencing
We chose representative CVV samples by county, year, and species and sent them to the National Microbiology Laboratory (Winnipeg, Manitoba, Canada) for full-genome sequencing. One PCR fragment was developed for the S segment, 3 for the M segment, and 5 for L segment (Table 2), and Sanger sequencing was performed by using BigDye version 3.1 on an ABI 3730X Analyzer L (both Thermofisher). Trace files were compiled by using SeqMan II (DNAStar, https://www.dnastar.com) to get consensus sequence for each segment. Alignments were generated by using ClustalW (https://www.clustal.org) and MEGA4 software (15). Phylogenetic trees were generated by using the maximum-likelihood method in Geneious version 11.1.5 (https://www.geneious.com) and PhyML (http://www.atgc-montpellier.fr) with the Jukes‒Cantor substitution model. Robustness of the nodes was evaluated by performing 500 bootstrap replicates. Trees were rooted with the Fort Sherman virus S, M, and L segments (GenBank accession nos. KX100130, KX100131, and KX100132). Mean nucleotide distances between and within CVV lineages were calculated by using MEGAX software (https://berkstech.psu.edu).
Mosquito Vector Competence
A colony of unknown generations of An. quadrimaculatus mosquitoes (Orlando strain) was obtained from BEI Resources (https://www.beiresources.org) (MRA-139) and were maintained at 27°C under standard rearing conditions (27 ± 1°C, 70% relative humidity, 12:12-h light:dark photoperiod) (16). Freshly propagated virus supernatant from infected Vero (African green monkey kidney) cultures were harvested at 48 h after infection (multiplicity of infection ≈1.0) and diluted 1:1 with defibrinated sheep blood and 2.5% sucrose mixture without freezing. In addition to undiluted supernatant, 10-fold dilutions from 1:10 to 1:10,000 were made in C6/36 maintenance medium (Eagle minimum essential medium containing 2% fetal bovine serum heat-inactivated with 0.5 g/L of sodium bicarbonate plus 0.1 mmol/L nonessential amino acids plus 100 U/mL penicillin/streptomycin) before being mixed 1:1 with defibrinated sheep blood and a final concentration of 2.5% sucrose. Female mosquitoes (3–5 days old) were deprived of sugar for 1–2 hours and allowed to feed on CVV-defibrinated sheep blood‒sucrose mixture for 30 min in a Hemotek membrane feeding system (Discovery Workshops, https://accrington.cylex-uk.co.uk) with a porcine sausage casing membrane at 37°C.
After feeding, females were anesthetized with CO2 and fully engorged mosquitoes were transferred to 0.6-liter cardboard containers and maintained with 10% sucrose at 27°C, 70% relative humidity¸ and a 12:12-h light: dark photoperiod. Infection, dissemination, and transmission assays were performed on days 6 and 15 after the infectious blood meal as described (17). On day 2 after feeding, because of the early time point, only infection and dissemination assays were performed. Dissemination rate is the proportion of mosquitoes with infected legs among infected mosquitoes; transmission rate is the proportion of mosquitoes with positive saliva among mosquitoes with disseminated infection. We compared infection, dissemination, and transmission rates among strains by using χ2 analysis, followed by Bonferroni corrections for multiple comparisons in GraphPad Prism version 7.05 (GraphPad Software, https://www.graphpad.com). We used a TaqMan real-time reverse transcription to detect CVV by using primers and probe targeting both lineage 1 and 2 (Table 1).
CVV Surveillance
Figure 2

Figure 2. Cache Valley virus infection rate, New York, USA, during 2000‒2016, calculated by using MLE, by year (A), mosquito species (B), New York regions (C), and combined mosquito species and years...
We sampled 1,842,352 female mosquitoes in 57,321 mosquito pools from 2000–2016, yielding a total of 255 CVV-positive pools. We compared MLE of prevalence by year (Figure 2, panel A), mosquito species (Figure 2, panel B), and regions (Figure 2, panel C). CVV activity fluctuated substantially during the 17-year sampling period. The highest estimates of prevalence were during 2003 (0.41, 95% CI 0.30‒0.53), 2010 (0.42, 95% CI 0.33‒0.54), and 2015 (0.52, 95% CI 0.41‒0.66). No CVV was detected during 2000, 2002, 2009, and 2016. Comparable CVV prevalence was measured in 2001 (0.08, 95% CI 0.03‒0.19), 2005 (0.06, 95% CI 0.02‒0.12), 2006 (0.06, 95% CI 0.02‒0.11), 2008 (0.13, 95% CI 0.07‒0.21), 2011 (0.08, 95% CI 0.04‒0.14), and 2013 (0.08, 95% CI 0.04‒0.08).
In addition, we calculated prevalence for 10 mosquito species that had the highest number of CVV isolations. The 5 mosquito species with the highest MLE were An. punctipennis (1.24), An. quadrimaculatus (0.53), Aedes cinereus (0.39), Ae. trivittatus (0.27), and Ae. sollicitans (0.18) (Figure 2, panel B). To show which mosquito genus was driving transmission of CVV in New York, we compared the combined top 5 mosquito MLEs of Aedes and Anopheles species. The prevalence for Ae. sollicitans, Ae. cinereus, and Ae. trivittatus mosquitoes was 0.29 compared with 0.28 for An. quadrimaculatus and An. punctipennis mosquitoes during 2000–2009 (Figure 2, panel D). During 2010–2016, the combined prevalence for An. quadrimaculatus and An. punctipennis mosquitoes increased significantly to 0.91 (p<0.05 by χ2 test), and prevalence for Ae. sollictans, Ae. cinereus, and Ae. trivitattus mosquitoes decreased to 0.21. Similar infection rates were observed throughout New York regions, except Long Island, where the rate was on average lower than those for the rest of the regions (0.04) (Figure 2, panel C).
Phylogenetic Analysis of CVV
Figure 3

Figure 3. Phylogenetic analysis of Cache Valley virus, New York, USA, 2000‒2016. Maximum-likelihood phylogenetic trees show complete nucleotide sequences of small (A), medium (B), and large (C) genome segments. Numbers at nodes...
We sequenced 48 CVV isolates representing various New York counties, hosts, and isolation dates and 3 isolates from Canada (Table 3). Most of the CVV isolates were from mosquitoes, except 4 that were isolated from 2 humans, 1 sheep, and 1 horse. Phylogenetic analysis of CVV confirmed 2 distinct lineages (lineages 1 and 2) (Figure 3). Lineage 1 contained all CVV strains obtained during 2001‒2010, and lineage 2 contained isolates obtained during 2011‒2016. Segment reassortment between M and S was observed in 4 samples, 3 from mosquito isolates (15041170, 15060131, and 15041084) and 1 from a human isolate (PA). All reassortants contain an lineage 1 L segment and lineage 2 S and M RNA segments. The 3 CVV strains isolated in Canada (1 isolate from a ewe placenta and 2 isolates from mosquito pools, all collected in Ontario during 2012) all grouped within lineage 2. There was no evidence of spatial clustering of clades within the S, M, and L segments, except the reassortants, which all came from western New York regions (Cattaraugus, Chautauqua, and Allegany Counties). Mean genetic distance calculated as the number of nucleotide substitutions per site between lineage was 0.040 for the S segment, 0.074 for the M segment, and 0.051 for the L segment (Table 4). On average, there were more nucleotide substitutions for the M segment (0.074) than for the S (0.040) and L (0.051) segments.
Mosquito Vector Competence
We conducted vector competence assays with An. quadrimaculatus mosquitoes for 2 lineage 1 (NY10, NY25), 3 lineage 2 (15350152, 15330577, and Hu2011), and 4 reassortant (15041084, PA, 15041170, and 15060131) strains to determine whether there were differences between the lineages or between strains in the same lineage and to address effects of reassortment. We also hoped to determine whether vector competence was a potential mechanism of displacement of lineage 1 (Tables 5, 6). Our results indicate that lineage 1 strains are generally less infectious in An. quadrimaculatus mosquitoes because they had a 50% infectious dose ≈0.5–1.0 log10 higher than that for lineage 2 (Table 5).
We also found decreased dissemination and transmission for lineage 1 strains of CVV compared with lineage 2 strains (p<0.05 by χ2 test) (Tables 5, 6). We observed that CVV disseminated efficiently in An. quadrimaculatus mosquitoes by 2 days postfeeding. All mosquitoes infected with lineage 2 strains had disseminated virus, and dissemination of lineage 1 strains was more variable (Tables 5, 6). In addition, An. quadrimaculatus mosquitoes are a competent vector for the lineage 2 human strain but not for the human reassortant (PA) strain (lineage 1 L RNA segment and lineage 2 S and M RNA segments), which had a low dissemination rate. Except for the PA strain, An. quadrimaculatus mosquitoes were able to transmit CVV at day 6 postfeeding on an artificial blood meal with a high viral titer (6.0–7.0 log10 PFU/mL). When mosquitoes were infected with a lower viral titer (4.0 log10 PFU/mL), the infection rate decreased from 95%–100% to 12% for lineage 1, from 100% to 28%–64% for lineage 2, and from 85%–100% to 24%–52% for reassortants (Tables 5, 6).
Consistent with the findings of Armstrong et al., who analyzed CVV strains from Connecticut (4), we identified substantial variability in CVV activity in New York during 2000–2016. In addition, in both states, CVV could be isolated from different mosquito genera, including Aedes, Anopheles, and Coquillettidia (6). In our study, the prevalence of CVV in An. punctipennis and An. quadrimaculatus mosquitoes during 2010–2016 (0.91) was higher than that during 2000–2009 (0.21). Although many mosquito species are apparently infected with CVV, our data and previous surveillance data for Connecticut (6) all point to Anopheles spp. mosquitoes driving virus activity.
At least 51 different viruses have been detected in Anopheles spp, including 14 viruses with potential to cause febrile disease if transmitted to humans or other vertebrates, such as o’nyong nyong virus, Venezuelan equine encephalitis virus, Western equine encephalitis virus, Sindbis virus, Semliki Forest virus, Rift Valley fever virus, West Nile virus, Japanese encephalitis virus, Wesselsbron virus, Tataguine virus, Batai virus, CVV, Tahyna virus, and Tensaw virus (18). However, only o’nyong nyong virus, which is closely related to chikungunya virus, is known to be consistently transmitted to vertebrates by Anopheles mosquitoes (19). Other studies supported potential roles of Anopheles mosquito species in the transmission of Rift Valley fever virus, Mayaro virus, Eastern equine encephalitis virus, and CVV (20–24). These data and our results confirmed that Anopheles mosquitoes have the potential to sustain transmission cycles of arboviruses. Additional studies are needed to elucidate their role in these cycles.
An. quadrimaculatus and An. punctipennis mosquitoes are mainly mammalian feeders in the northeastern United States, and white-tailed deer is the most commonly identified vertebrate host (25). Both mosquito species bite outdoors throughout the night and show higher activity at dusk and dawn and resting outdoors (26,27). In New York, white-tailed deer tested for CVV antibodies showed infection rates of 25.7% (28). White-tailed deer have been identified as the principal reservoir and amplification hosts for CVV, and their overabundance and availability for both Anopheles mosquitoes species that are frequently infected by the virus in nature (6,27–30) could partially explain the increase of CVV activity in Anopheles spp. observed in our study.
Early phylogenetic analysis of CVV strains from United States and Canada showed only a single lineage (31,32). Armstrong et al. reported emergence of a new lineage of CVV in Connecticut during 2010, displacement of lineage 1 by 2014, and no evidence of genome reassortment (4). Our phylogenetic analysis confirmed that the displacement of CVV lineage 1 was widespread in the region and throughout eastern Canada because the CVV lineage 2 was responsible for several outbreaks of fetal malformation disease in Ontario and Quebec sheep flocks during 2012 and 2013 (M.A. Drebot, unpub. data). Furthermore, we demonstrated that An. quadrimaculatus mosquitoes are a competent vector for both CVV lineages and reassortants. The differential susceptibility between lineage 1 and lineage 2 suggest that An. quadrimaculatus mosquitoes might be actively involved in lineage 1 displacement in the northeast United States and can potentially increase the risk for spillover to humans in the region because lineage 2 is more infectious and more readily transmitted.
We isolated 4 reassortant strains that contained lineage 1 L segments and lineage 2 S and M RNA segments, and all came from counties in western New York. Reassortment is an evolutionary mechanism of segmented RNA viruses to exchange genetic information during co-infection of cells, which generates new genotypes and phenotypes (33,34). During reassortment, entire genes are exchanged among different viral strains or species by the swapping of segments, which confer major fitness advantages or disadvantages to the progeny virus (34). In the family Peribunyaviridae, reassortment events have occurred between virus lineages. Intraspecies, interlineage reassortment events were reported for Rift Valley fever virus, a phlebovirus and a mosquitoborne zoonotic virus that affects domestic animals and humans (35), and also for Crimean-Congo hemorrhagic fever virus (33,36,37), a highly infectious orthonairovirus transmitted by Hyalomma spp. ticks. Furthermore, interspecies reassortment also occurs. For example, reassortment among Bunymawera serogroup viruses has been documented with Ngari virus and Potosi virus (38–41), among others. In addition, although segment reassortment among California serogroup viruses is infrequent (42), evidence of reassortment has been documented (43,44).
Earlier studies had demonstrated that genetic reassortment between members of the family Peribunyaviridae can occur in vitro in mosquito and mammal cells and in vivo in mosquitoes during a mixed infection and can produce viable new strains with major phenotypic changes in terms of infectivity and pathogenicity (38–40,42,45–47). Furthermore, the phenomenon of superinfection resistance might promote opportunities for segment reassortment between more distantly related viruses. However, co-infection by closely related viruses can occur only in cases in which the second virus infects rapidly after the first virus and before superinfection resistance becomes effective (38). In our study, 3 CVV reassortants were isolated from mosquitoes and 1 was isolated from a human, and all contained the CVV lineage 1 L segment and CVV lineage 2 S and M segments. In addition, reassortant mosquito isolates that contained the L RNA segment with CVV lineage 1 were more infectious for An. quadrimaculatus mosquitoes than the lineage 1 strains, suggesting a probable role of the S or M RNA segments of lineage 2 strain in mosquito infectivity.
The vector competence of Ae. albopictus mosquitoes for Potosi virus and the susceptibility of An. gambiae Giles mosquitoes for Ngari virus has been demonstrated (48,49). Among the reassortant strains tested in our study, only the human reassortant strain was not transmitted by An. quadrimaculatus mosquitoes despite persistent infection. This difference in phenotype was probably not caused by the viral titer in the infectious blood meal because the titer was only ≈0.5 log10 lower for the human reassortant strain. We suspect that difference might be caused by other factors involving the virus strain and mosquito species used in our study. Addressing the potential mechanisms involved in differential vector competence phenotypes observed in An. quadrimaculatus mosquitoes and evaluating the role of strain variation in host competence and pathogenicity will help to clarify the consequences of genetic variation and displacement of CVV.
Dr. Dieme is research scientist at the Wadsworth Center, New York State Department of Health, Slingerlands, NY. Her primary research interest is vector‒pathogen interactions, especially in mosquito infections.
Acknowledgments
We thank the New York State Department of Health, the Wadsworth Center Media, and the Tissue Core Facility for providing cells and media for these studies; the New York State Arbovirus Laboratory insectary staff for providing assistance with rearing and experimentation; and Maya Andonova and Kimberly Holloway for providing technical assistance.
This study was supported by cooperative agreement U01CK000509 from the Centers for Disease Control and Prevention.
References
- Virus Taxonomy. 2018b release EC 50, Washington, DC, July 2018 Email ratification February 2019 (MSL #34) [cited 2021 Oct 16]. https://talk.ictvonline.org/files/master-species-lists/m/msl/8266
- Elliott RM. Molecular biology of the Bunyaviridae. J Gen Virol. 1990;71:501–22. DOIPubMedGoogle Scholar
- Dunlop JI, Szemiel AM, Navarro A, Wilkie GS, Tong L, Modha S, et al. Development of reverse genetics systems and investigation of host response antagonism and reassortment potential for Cache Valley and Kairi viruses, two emerging orthobunyaviruses of the Americas. PLoS Negl Trop Dis. 2018;12:
e0006884 . DOIPubMedGoogle Scholar - Armstrong PM, Andreadis TG, Anderson JF. Emergence of a new lineage of Cache Valley virus (Bunyaviridae: Orthobunyavirus) in the Northeastern United States. Am J Trop Med Hyg. 2015;93:11–7. DOIPubMedGoogle Scholar
- Waddell L, Pachal N, Mascarenhas M, Greig J, Harding S, Young I, et al. Cache Valley virus: A scoping review of the global evidence. Zoonoses Public Health. 2019;66:739–58. DOIPubMedGoogle Scholar
- Andreadis TG, Armstrong PM, Anderson JF, Main AJ. Spatial-temporal analysis of Cache Valley virus (Bunyaviridae: Orthobunyavirus) infection in anopheline and culicine mosquitoes (Diptera: Culicidae) in the northeastern United States, 1997-2012. Vector Borne Zoonotic Dis. 2014;14:763–73. DOIPubMedGoogle Scholar
- Newhouse VF, Siverly RES. St. Louis encephalitis virus from mosquitoes in southwestern Indiana, 1964. J Med Entomol. 1966;3:340–2. DOIPubMedGoogle Scholar
- Reiter P. A portable battery-powered trap for collecting gravid Culex mosquitoes. Mosq News. 1983;43:496–8.
- Means RG. Mosquitoes of New York, Part II: genera of Culicidae other than Aedes occuring in New York. New York State Museum, 1987;430B [cited 2021 Oct 16]. http://www.nysm.nysed.gov/staff-publications/mosquitoes-new-york-part-ii-genera-culicidae-other-ae
- Kauffman EB, Jones SA, Dupuis AP II, Ngo KA, Bernard KA, Kramer LD. Virus detection protocols for west nile virus in vertebrate and mosquito specimens. J Clin Microbiol. 2003;41:3661–7. DOIPubMedGoogle Scholar
- Oliver J, Lukacik G, Kokas J, Campbell SR, Kramer LD, Sherwood JA, et al. Twenty years of surveillance for Eastern equine encephalitis virus in mosquitoes in New York State from 1993 to 2012. Parasit Vectors. 2018;11:362. DOIPubMedGoogle Scholar
- Yang Y, Qiu J, Snyder-Keller A, Wu Y, Sun S, Sui H, et al. Fatal Cache Valley virus meningoencephalitis associated with rituximab maintenance therapy. Am J Hematol. 2018;93:590–4. DOIPubMedGoogle Scholar
- Nguyen NL, Zhao G, Hull R, Shelly MA, Wong SJ, Wu G, et al. Cache valley virus in a patient diagnosed with aseptic meningitis. J Clin Microbiol. 2013;51:1966–9. DOIPubMedGoogle Scholar
- Ngo KA, Maffei JG, Dupuis AP II, Kauffman EB, Backenson PB, Kramer LD. Isolation of Bunyamwera serogroup viruses (Bunyaviridae, Orthobunyavirus) in New York state. J Med Entomol. 2006;43:1004–9.PubMedGoogle Scholar
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–9. DOIPubMedGoogle Scholar
- Ciota AT, Matacchiero AC, Kilpatrick AM, Kramer LD. The effect of temperature on life history traits of Culex mosquitoes. J Med Entomol. 2014;51:55–62. DOIPubMedGoogle Scholar
- Ciota AT, Bialosuknia SM, Zink SD, Brecher M, Ehrbar DJ, Morrissette MN, et al. Effects of Zika Virus strain and Aedes mosquito species on vector competence. Emerg Infect Dis. 2017;23:1110–7. DOIPubMedGoogle Scholar
- Nanfack Minkeu F, Vernick KD. A systematic review of the natural virome of Anopheles mosquitoes. Viruses. 2018;10:10. DOIPubMedGoogle Scholar
- Carissimo G, Pain A, Belda E, Vernick KD. Highly focused transcriptional response of Anopheles coluzzii to O’nyong nyong arbovirus during the primary midgut infection. BMC Genomics. 2018;19:526. DOIPubMedGoogle Scholar
- Dieme C, Ciota AT, Kramer LD. Transmission potential of Mayaro virus by Aedes albopictus, and Anopheles quadrimaculatus from the USA. Parasit Vectors. 2020;13:613. DOIPubMedGoogle Scholar
- Blackmore CG, Blackmore MS, Grimstad PR. Role of Anopheles quadrimaculatus and Coquillettidia perturbans (Diptera: Culicidae) in the transmission cycle of Cache Valley virus (Bunyaviridae: Bunyavirus) in the midwest, USA. J Med Entomol. 1998;35:660–4. DOIPubMedGoogle Scholar
- Brustolin M, Pujhari S, Henderson CA, Rasgon JL. Anopheles mosquitoes may drive invasion and transmission of Mayaro virus across geographically diverse regions. PLoS Negl Trop Dis. 018;12:e0006895.
- Moncayo AC, Edman JD, Turell MJ. Effect of eastern equine encephalomyelitis virus on the survival of Aedes albopictus, Anopheles quadrimaculatus, and Coquillettidia perturbans (Diptera: Culicidae). J Med Entomol. 2000;37:701–6. DOIPubMedGoogle Scholar
- Nepomichene TNJJ, Raharimalala FN, Andriamandimby SF, Ravalohery J-P, Failloux A-B, Heraud J-M, et al. Vector competence of Culex antennatus and Anopheles coustani mosquitoes for Rift Valley fever virus in Madagascar. Med Vet Entomol. 2018;32:259–62. DOIPubMedGoogle Scholar
- Molaei G, Farajollahi A, Armstrong PM, Oliver J, Howard JJ, Andreadis TG. Identification of bloodmeals in Anopheles quadrimaculatus and Anopheles punctipennis from eastern equine encephalitis virus foci in northeastern U.S.A. Med Vet Entomol. 2009;23:350–6. DOIPubMedGoogle Scholar
- Molaei G, Andreadis TG, Armstrong PM, Diuk-Wasser M. Host-feeding patterns of potential mosquito vectors in Connecticut, U.S.A.: molecular analysis of bloodmeals from 23 species of Aedes, Anopheles, Culex, Coquillettidia, Psorophora, and Uranotaenia. J Med Entomol. 2008;45:1143–51. DOIPubMedGoogle Scholar
- McMillan JR, Armstrong PM, Andreadis TG. Patterns of mosquito and arbovirus community composition and ecological indexes of arboviral risk in the northeast United States. PLoS Negl Trop Dis. 2020;14:
e0008066 . DOIPubMedGoogle Scholar - Dupuis AP, Prusinski MA, Russell A, O’Connor C, Maffei JG, Oliver J, et al. Serologic survey of mosquito-borne viruses in hunter-harvested white-tailed deer (Odocoileus virginianus), New York State. Am J Trop Med Hyg. 2020;104:593–603. DOIPubMedGoogle Scholar
- Whitney E, Roz AP, Rayner GA, Deibel R. Serologic survey for arbovirus activity in deer sera from nine counties in New York State. Wildl Dis. 1969;5:392–7. DOIPubMedGoogle Scholar
- Seggos B. Deer management in urban and suburban New York : a report to the New York State Senate and Assembly, 2018 [cited 2021 Oct 16]. https://www.dec.ny.gov/docs/wildlife
- Blitvich BJ, Loroño-Pino MA, Garcia-Rejon JE, Farfan-Ale JA, Dorman KS. Nucleotide sequencing and serologic analysis of Cache Valley virus isolates from the Yucatan Peninsula of Mexico. Virus Genes. 2012;45:176–80. DOIPubMedGoogle Scholar
- Pabbaraju K, Ho KC, Wong S, Fox JD, Kaplen B, Tyler S, et al. Surveillance of mosquito-borne viruses in Alberta using reverse transcription polymerase chain reaction with generic primers. J Med Entomol. 2009;46:640–8. DOIPubMedGoogle Scholar
- Zhou Z, Deng F, Han N, Wang H, Sun S, Zhang Y, et al. Reassortment and migration analysis of Crimean-Congo haemorrhagic fever virus. J Gen Virol. 2013;94:2536–48. DOIPubMedGoogle Scholar
- McDonald SM, Nelson MI, Turner PE, Patton JT. Reassortment in segmented RNA viruses: mechanisms and outcomes. Nat Rev Microbiol. 2016;14:448–60. DOIPubMedGoogle Scholar
- Bird BH, Khristova ML, Rollin PE, Ksiazek TG, Nichol ST. Complete genome analysis of 33 ecologically and biologically diverse Rift Valley fever virus strains reveals widespread virus movement and low genetic diversity due to recent common ancestry. J Virol. 2007;81:2805–16. DOIPubMedGoogle Scholar
- Goedhals D, Bester PA, Paweska JT, Swanepoel R, Burt FJ. Next-generation sequencing of southern African Crimean-Congo haemorrhagic fever virus isolates reveals a high frequency of M segment reassortment. Epidemiol Infect. 2014;142:1952–62. DOIPubMedGoogle Scholar
- Hewson R, Gmyl A, Gmyl L, Smirnova SE, Karganova G, Jamil B, et al. Evidence of segment reassortment in Crimean-Congo haemorrhagic fever virus. J Gen Virol. 2004;85:3059–70. DOIPubMedGoogle Scholar
- Briese T, Calisher CH, Higgs S. Viruses of the family Bunyaviridae: are all available isolates reassortants? Virology. 2013;446:207–16. DOIPubMedGoogle Scholar
- Groseth A, Weisend C, Ebihara H. Complete genome sequencing of mosquito and human isolates of Ngari virus. J Virol. 2012;86:13846–7. DOIPubMedGoogle Scholar
- Dutuze MF, Nzayirambaho M, Mores CN, Christofferson RC. A review of Bunyamwera, Batai, and Ngari viruses: understudied orthobunyaviruses with potential one health implications. Front Vet Sci. 2018;5:69. DOIPubMedGoogle Scholar
- Briese T, Kapoor V, Lipkin WI. Natural M-segment reassortment in Potosi and Main Drain viruses: implications for the evolution of orthobunyaviruses. Arch Virol. 2007;152:2237–47. DOIPubMedGoogle Scholar
- Hughes HR, Lanciotti RS, Blair CD, Lambert AJ. Full genomic characterization of California serogroup viruses, genus Orthobunyavirus, family Peribunyaviridae including phylogenetic relationships. Virology. 2017;512:201–10. DOIPubMedGoogle Scholar
- Urquidi V, Bishop DH. Non-random reassortment between the tripartite RNA genomes of La Crosse and snowshoe hare viruses. J Gen Virol. 1992;73:2255–65. DOIPubMedGoogle Scholar
- Reese SM, Blitvich BJ, Blair CD, Geske D, Beaty BJ, Black WC IV. Potential for La Crosse virus segment reassortment in nature. Virol J. 2008;5:164. DOIPubMedGoogle Scholar
- Endres MJ, Griot C, Gonzalez-Scarano F, Nathanson N. Neuroattenuation of an avirulent bunyavirus variant maps to the L RNA segment. J Virol. 1991;65:5465–70. DOIPubMedGoogle Scholar
- Gerrard SR, Li L, Barrett AD, Nichol ST. Ngari virus is a Bunyamwera virus reassortant that can be associated with large outbreaks of hemorrhagic fever in Africa. J Virol. 2004;78:8922–6. DOIPubMedGoogle Scholar
- Cheng LL, Rodas JD, Schultz KT, Christensen BM, Yuill TM, Israel BA. Potential for evolution of California serogroup bunyaviruses by genome reassortment in Aedes albopictus. Am J Trop Med Hyg. 1999;60:430–8. DOIPubMedGoogle Scholar
- Mitchell CJ, Smith GC, Miller BR. Vector competence of Aedes albopictus for a newly recognized Bunyavirus from mosquitoes collected in Potosi, Missouri. J Am Mosq Control Assoc. 1990;6:523–7.PubMedGoogle Scholar
- Odhiambo C, Venter M, Chepkorir E, Mbaika S, Lutomiah J, Swanepoel R, et al. Vector competence of selected mosquito species in Kenya for Ngari and Bunyamwera viruses. J Med Entomol. 2014;51:1248–53. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleOriginal Publication Date: January 04, 2022
1These authors contributed equally to this article.
Table of Contents – Volume 28, Number 2—February 2022
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Constentin Dieme, New York State Department of Health, 5668 State Farm Rd, Slingerlands, NY 12159, USA
Top