Volume 28, Number 2—February 2022
Dispatch
SARS-CoV-2 B.1.619 and B.1.620 Lineages, South Korea, 2021
Cite This Article
Citation for Media
Abstract
We report the rapid emergence of severe acute respiratory syndrome coronavirus 2 lineages B.1.619 and B.1.620 in South Korea. The surge in frequency in a relatively short time emphasizes the need for ongoing monitoring for new lineages to track potential increases in transmissibility and disease severity and reductions in vaccine efficacy.
Since the emergence of coronavirus disease (COVID-19) in December 2019, ≈2 million genomes of SARS-CoV-2 have been sequenced worldwide, revealing that the virus is continuously mutating (1). The mutations of SARS-CoV-2 spike protein must be monitored because of its vital role in attaching to the host cell-surface receptor, angiotensin-converting enzyme 2 (ACE2) (2), which increases its infectivity (3).
As of May 31, 2021, the World Health Organization (WHO) reported the appearance of several variants of concern (VOCs) whose characteristics have serious implications on public health: Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1), and Delta (B.1.617.2) (4). These variants arose from changes in the spike protein, especially in the receptor-binding domain (RBD). The RBD plays an important role in direct interaction with human ACE2; the 4 variants contain >1 of the specific substitutions (K417N, L452R, T478K, E484K, and N501Y) that affect viral fitness and transmissibility. These variants possess substantially higher transmissibility, evade immunity, increase disease severity, reduce vaccine efficacy, and escape diagnostic detection (5). WHO designated Lambda (C.37) as a variant of interest (VOI) on June 14, 2021, and Mu (B.1.621) as a VOI on August 30, 2021. These variants were considered likely to become highly transmissible and evade vaccine protection, thus threatening South America, where they were first identified (Acevedo et al., unpub. data, https://doi.org/10.1101/2021.06.28.21259673) (6).
Genomic surveillance and open data sharing of viral genome sequences have enhanced near–real-time detection, comparison, and tracking of SARS-CoV-2 variants (1). The Korea Disease Control and Prevention Agency (KDCA) has been conducting whole-genome sequencing (WGS) of SARS-CoV-2 in South Korea since the beginning of the pandemic; targeted sequencing of the spike protein is being implemented to strengthen new variant monitoring. Lineage distribution analysis in South Korea indicated that, from its discovery in March 2020 until January 2021, the B.1.497 lineage was predominant in domestic cases (7,8). B.1.497 formed 1 of 4 major clusters in South Korea but was the only one that had expanded and predominantly circulated in the country (9). Sequence analysis indicated that it was originally clustered with North American viruses; additional genetic mutations in this cluster are H1113Y, T2408N, P4223S, and A5770S in open reading frame 1ab (ORF1ab) and Q52H, A222V, E556K, T716I, and A1070V in the spike protein. However, changes have been observed in lineage distribution since March 2021; increases in B.1.619 and B.1.620 are of note. We report the sudden emergence of these 2 lineages harboring the E484K mutation in the spike protein, which rapidly outcompeted the existing variants in South Korea.
We collected nasopharyngeal and oropharyngeal swab samples from patients with SARS-CoV-2 cases confirmed by real-time reverse transcription PCR (rRT-PCR). We prepared WGS libraries using QIAseq SARS-CoV-2 Primer Panel and QIAseq FX DNA Library UDI Kit (QIAGEN, https://www.qiagen.com) and sequenced them on MiSeq (Illumina, https://www.illumina.com) with 2 × 150 bp using MiSeq reagent kit version 2 (Illumina). For phylogenetic tree analysis, we aligned whole genomic sequences using MAFFT version 7 (https://mafft.cbrc.jp/alignment/server), inferred maximum-likelihood phylogenetic trees with FastTree version 2.1.9 (http://www.microbesonline.org/fasttree), and visualized using Interactive Tree of Life version 5 (https://itol.embl.de). The sequences have been uploaded to the GISAID EpiCoV database (https://www.gisaid.org).
Figure 1
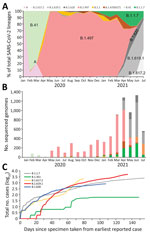
Figure 1. Investigation of SARS-CoV-2 B.1.619 and B.1.620 lineages, South Korea, 2021. A) Distribution of 7 SARS-CoV-2 lineages in domestic cases over time. Data are shown for lineages A, B.41, B.1.497, B.1.1.7...
We obtained WGS of 9,554 SARS-CoV-2 as of July 21, 2021, representing >5% of the total reported positive cases during this period. Specifically, we obtained WGS for 6.2% of the total positive cases during April–July, when the number of infections by B.1.619 and B.1.620 increased sharply. Of those, 7,585 were domestic cases. As described previously (7), the A and B.41 lineages were the most prevalent at the beginning of the pandemic in South Korea. However, B.1.497 (formerly known as B.1.3.1) gained predominance in South Korea after its emergence in March 2020 (8,9). The lineage distribution has been changing since January 2021 as VOCs have emerged. The earliest recorded Alpha variant in South Korea was identified on December 22, 2020; its prevalence increased to 22.0% as of June 2021 (Figure 1). Simultaneously, prevalence rates of B.1.619 and B.1.620 increased rapidly; prevalence was 55.4% in March 2021 and 11.5% in June 2021. The increase in the prevalence of these 2 lineages was rapid; it reached 67% in June, but decreased slightly when the Delta variant emerged in July. The prevalence of B.1.497 decreased from 94.3% to 0.9% in June 2021.
Figure 2
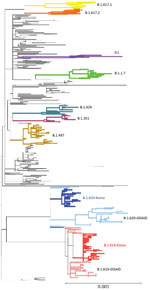
Figure 2. Phylogenetic analysis of severe acute respiratory syndrome coronavirus 2 sequences, South Korea. A total of 457 sequences were used to construct the tree, including 37 sequences of B.1.619 lineage and...
B.1.619 and B.1.620 were identified in imported cases in South Korea in 2021; B.1.619 in a case-patient from Cameroon in February and B.1.620 in cases from Kenya and Malawi in March. The phylogenetic analysis of SARS-CoV-2 sequences isolated in South Korea showed that B.1.619 and B.1.620 were distinct from those in countries in Europe (Figure 2); this finding indicates 1 or very few introduction events for B.1.619 and B.1.620 strains into South Korea, from which strains then spread rapidly. South Korea B.1.619 has been reclassified as B.1.619.1, which has an additional mutation in ORF1ab (K3929R) (10).
B.1.620 was prevalent in central Africa and later spread to Europe and the United States through travelers (10). We were unable to find previous research on B.1.619 in the literature; however, we assumed that B.1.619 was a prevalent strain in central Africa and later spread to Europe because it has been identified in central Africa, according to information from GISAID.
The B.1.619 and B.1.620 lineages have several characteristic spike protein mutations (Table 1); the E484K mutation, which is present in both Beta and Gamma variants and has been identified as an escape mutation (11), is the only shared mutation in both lineages. The mutations in the spike protein, specifically in the RBD, have a strong influence on SARS-CoV-2 pathogenesis; B.1.619 has additional N440K mutations in the RBD and B.1.620 has S447N substitutions. The S477N mutation may evade antibody-mediated immunity (12) and increase RBD affinity for ACE2 (13). In addition, the N440K mutation might confer resistance to monoclonal antibodies and enhance binding affinity to the ACE2 receptor (13,14). We observed no other specific mutation in the spike protein in B.1.619. In contrast, B.1.620 carries several mutations and deletions, previously observed individually in VOCs and VOIs (Table 1). The HV69/70Δ, Y144Δ, P681H, and D1118H mutations in the spike protein have been found in the Alpha variant, whereas the LAL242/243/244Δ mutation has been found in the Beta variant. Previously, we found that B.1.619 and 620 have no inhibitory effect on the neutralizing activity in vaccinated or convalescent persons (S.J. Oh et al., unpub. data). However, the combined effect of these mutations on viral pathogenicity and transmissibility needs to be elucidated.
Continuous monitoring of mutations is essential to track potential vaccine efficacy reduction, increased transmissibility, and disease severity. The transmissibility of B.1.619 and B.1.620 and their likelihood to cause more severe infections are not yet confirmed. Preliminary data show that patients who recovered from non-VOC/VOI and vaccinated persons have sufficient neutralizing capacity against these lineages (Table 2). The transmissibility and immune escape of these strains must be investigated further. Continuous genomic surveillance supporting public health response is required to overcome the COVID-19 pandemic.
Dr. Ae Kyung Park is a staff scientist at the Korea Disease Control and Prevention Agency. Her research interests include molecular epidemiology of pathogens, and next-generation sequencing.
Acknowledgments
We thank the authors of the originating laboratories and the submitting laboratories who have deposited and shared genome data on GISAID’s EpiCoV (https://www.gisaid.org) database.
This work was supported by a grant from the Korea Disease Control and Prevention Agency (grant no. 4800-4837-301).
References
- Shu Y, McCauley J. GISAID: Global initiative on sharing all influenza data - from vision to reality. Euro Surveill. 2017;22:30494. DOIPubMedGoogle Scholar
- Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol. 2020;5:562–9. DOIPubMedGoogle Scholar
- Zhang L, Jackson CB, Mou H, Ojha A, Peng H, Quinlan BD, et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat Commun. 2020;11:6013. DOIPubMedGoogle Scholar
- World Health Organization. Weekly epidemiological update on COVID-19—1 June 2021. Edition 42. 2021 [cited 2021 Dec 14]. https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---1-june-2021
- European Centre for Disease Prevention and Control. SARS-CoV-2 variants of concern as of 3 June 2021. 2021 [cited 2021 Dec 21]. https://ecdc.europa.eu/en/covid-19/variants-concern.
- Laiton-Donato K, Franco-Muñoz C, Álvarez-Díaz DA, Ruiz-Moreno HA, Usme-Ciro JA, Prada DA, et al. Characterization of the emerging B.1.621 variant of interest of SARS-CoV-2. Infect Genet Evol. 2021;95:
105038 . DOIPubMedGoogle Scholar - Park AK, Kim IH, Kim J, Kim JM, Kim HM, Lee CY, et al. Genomic surveillance of SARS-CoV-2: distribution of clades in South Korea in 2020. Osong Public Health Res Perspect. 2021;12:37–43. DOIPubMedGoogle Scholar
- Rambaut A, Holmes EC, O’Toole Á, Hill V, McCrone JT, Ruis C, et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol. 2020;5:1403–7. DOIPubMedGoogle Scholar
- Kwon J-H, Kim J-M, Lee D-H, Park AK, Kim I-H, Kim D-W, et al. Genomic epidemiology reveals the reduction of the introduction and spread of SARS-CoV-2 after implementing control strategies in Republic of Korea, 2020. Virus Evol. 2021;7:veab077. DOIGoogle Scholar
- Latif AA, Mullen JL, Alkuzweny M, Tsueng G, Cano M, Haag E, et al. Center for Viral Systems Biology. B.1.619.1 lineage report. 2021 [cited 2021 Dec 14]. https://outbreak.info/situation-reports?pango=B.1.619.1
- Harvey WT, Carabelli AM, Jackson B, Gupta RK, Thomson EC, Harrison EM, et al.; COVID-19 Genomics UK (COG-UK) Consortium. SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol. 2021;19:409–24. DOIPubMedGoogle Scholar
- Liu Z, VanBlargan LA, Bloyet LM, Rothlauf PW, Chen RE, Stumpf S, et al. Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe. 2021;29:477–488.e4. DOIPubMedGoogle Scholar
- Starr TN, Greaney AJ, Hilton SK, Ellis D, Crawford KHD, Dingens AS, et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell. 2020;182:1295–1310.e20. DOIPubMedGoogle Scholar
- Weisblum Y, Schmidt F, Zhang F, DaSilva J, Poston D, Lorenzi JC, et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. eLife. 2020;9:
e61312 . DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleOriginal Publication Date: January 04, 2022
Table of Contents – Volume 28, Number 2—February 2022
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Eun-Jin Kim, Ph.D. Division of Emerging Infectious Diseases, Bureau of Infectious Diseases Diagnosis Control, Korea Disease Control and Prevention Agency (KDCA), 187 Osongsaengmyeong 2-ro, Osong-eup, Heungdeok-gu, Cheongju-si, Chungcheongbuk-do 28159, South Korea; e-mail:ekim@korea.kr
Top