Volume 30, Number 6—June 2024
Dispatch
Evolution and Antigenic Differentiation of Avian Influenza A(H7N9) Virus, China
Cite This Article
Citation for Media
Abstract
We characterized the evolution and molecular characteristics of avian influenza A(H7N9) viruses isolated in China during 2021–2023. We systematically analyzed the 10-year evolution of the hemagglutinin gene to determine the evolutionary branch. Our results showed recent antigenic drift, providing crucial clues for updating the H7N9 vaccine and disease prevention and control.
From early 2013 through October 2017, a total of 5 outbreaks of avian influenza A(H7N9) virus infection occurred, resulting in 616 human deaths (1). In particular, the fifth wave of the epidemic saw a substantial increase in human fatalities. By late 2017, a total of 1,568 laboratory-confirmed cases of H7N9 virus infection in humans had been reported according to International Health Regulations guidelines (https://www.who.int/emergencies/disease-outbreak-news/item/26-october-2017-ah7n9-china-en). The rapid emergence, prevalence, and pandemic potential of H7N9 virus were suddenly of great concern. Since 2017, low-pathogenicity avian influenza H7N9 virus transformed into the highly pathogenic avian influenza (HPAI) A(H7N9) virus (2–5). In response, China initiated a large-scale vaccination program in the poultry industry, effectively limiting the H7N9 epidemic. Although no human H7N9 infections have been reported since February 2019, the virus is still circulating in poultry, particularly in laying hens, and remains a potential threat to poultry industry and public health (6–8). Furthermore, since 2017, the H7N9 virus has undergone multiple instances of antigenic drift to evade immune pressure from vaccines (9–11). We investigated the genetic evolution and antigenic differentiation of the H7N9 virus in China to provide information to better control the epidemic, ensure the safety of the poultry industry, and protect public health.
Figure 1

Figure 1. Phylogenetic analysis of evolution and antigenic differentiation of avian influenza A(H7N9) virus, China. Colors in columns at left show locations, timeframes, hosts, and pathogenicity of virus strains. The maximum-likelihood phylogenetic...
Through continuous monitoring of markets and breeding farms in several provinces, we successively isolated 23 H7N9 viruses. Using the sequences of those viruses and a reference sequence from the GISAID database (12), we conducted a phylogenetic analysis to study the evolution of H7N9 virus over the past decade (Figure 1).
We rooted the maximum-likelihood phylogenetic tree with A/Anhui/1/2013 and identified the branches as Group.y.0. During 2013–2017, the 5 low-pathogenicity avian influenza H7N9 virus waves formed Group.y.0–Group.y.2 branch. The first wave was mainly prevalent in the Yangtze River Delta. In 2014, the second wave spread to the Pearl River Delta and gradually expanded to all parts of the country in the subsequent 3 waves. Around 2017, or even as early as mid-2016, researchers successfully isolated an HPAI variant of H7N9 virus (6). That variant was found to contain alkaline amino acids inserted into the cleavage site of the hemagglutinin protein (6,13). The discovery of that variant in live poultry markets in Guangdong Province indicated an increased pathogenicity to poultry and potentially posed a greater threat to human health. We found that those HPAI H7N9 virus variants clustered within the Group.y.2.1 branch and its subordinate branch.
After implementation of a universal immunization program in 2017, the H7N9 virus outbreak was effectively controlled. However, some H7N9 viruses evolved to evade the vaccine. Those viruses continued to evolve and formed a new branch, Group.y.2.2, which is mainly found in northern China (Figure 1). Further investigation revealed that H7N9 virus is prevalent in the Bohai Rim region. Of note, we found no great differences in geographic, temporal, or host distribution between the 2 newly differentiated branches, Group.y.2.3 (A/Chicken/Hebei/1009/2020-like) and Group.y.2.4 (A/Chicken/Yunnan/1001/2021-like) (Figure 1). Those branches showed an average of 2.41% pairwise nucleotide distances between 2021 and 2023. That finding suggests that the evolutionary differences between those clades might not be influenced by geographic isolation, period, or host species, but rather by the adaptation of a new virus to natural selection. Positive selection pressure, which encourages mutations that contribute to the virus’ adaptation to the environment, can play a role in viral evolution. Our analysis confirmed that an increase in positive selection pressure in H7N9 virus occurred at some sites after 2017 (Appendix Table 1).
Figure 2
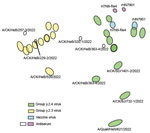
Figure 2. Antigenic map of avian influenza A(H7N9) virus, China, 2021–2023. The map was plotted using hemagglutinin inhibition assay results of 26 antigens (green, blue, and yellow dots), serum from 2 vaccine...
To examine whether mutation and evolution of H7N9 viruses are a result of antigenic drift and discontinuous variation, we used serologic methods to assess the antigenicity of the more evolved viruses from different clades (Appendix Table 2). Using the H7-Re4 and rHN7901 vaccine viruses for comparison, we found a weak cross-reaction titer between the vaccine viruses and the epidemic viruses in Group.y.2.3 (Table). The antigenic map demonstrated that the Group.y.2.3 viruses were distantly located from the vaccine serum (Figure 2), implying a consistent antigenic drift and greater antigenic divergence from Group.y.2.4 viruses. However, the distance between the vaccine virus and certain Group.y.2.4 viruses was relatively close (Figure 2), suggesting minimal differences. Furthermore, some Group.y.2.4 viruses, including A/Chicken/BJ/732-1/2022 and A/Quail/HeN/621/2022, both originating from northern China, also exhibited antigenic drift.
Changes in antigenicity often are caused by accumulation of amino acid mutations in antigenic sites. Therefore, we compared virus sequences and observed that the cleavage sites were KRKRTAR↓GLF or KRKRIAR↓GLF, both of which exhibited the characteristics of HPAI viruses. However, we noted no substantial differences between Group.y.2.3 and Group.y.2.4 at positions 86, 129, 134, 141, 145, 148, 151, 159, 208, 284, and 319 of H7 (Appendix Table 3). Those findings demonstrated the high genetic diversity of the H7N9 virus. Except for position 208 in H7, all sites were antigenic sites, and positions 141, 145, and 148 were both antigenic sites and receptor-binding sites. For the Group.y.2.4 branch, we compared the hemagglutinin 1 peptide of the vaccine viruses against antigenically distant viruses A/Chicken/SD/1301-2/2022, A/Chicken/HeB/B363-4/2022, A/Chicken/BJ/B732-1/2022, and A/Quail/HeN/621/2022. We observed different amino acids that could affect H7N9 virus antigenicity (Appendix Table 4). Among the analyzed viruses, A/Quail/HeN/621/2022 exhibited the highest number of mutations compared with the vaccine viruses, followed by A/Chicken/BJ/B732-1/2022 and A/Chicken/HeB/B363-4/2022; A/Chicken/SD/1401-2/2022 displayed the fewest mutations. Moreover, the previously reported Q226L and G228S sites of H3 viruses (Appendix Table 5), which have the potential to enhance mammalian adaptation, remained unchanged in all H7N9 viruses. Those sites still showed a preference for avian receptors, except A/Quail/HeN/621/2022, which mutated to P at position 160. All V125T H3 sites were replaced, indicating that the receptor-binding capacity and immune escape of the virus might be affected, making the virus more compatible with avian receptors (14,15).
This study explored the evolution and antigenic differentiation characteristics of H7N9 virus over the past decade through continuous monitoring and selection of representative sequences from all publicly available H7N9 virus sequences. However, our research still had certain limitations, and further investigation is needed to understand the relationship between the evolution of viruses under positive selection pressure and the underlying cause of antigenic variation.
In summary, influenza A viruses are highly prone to mutation and evolution, making the H7N9 virus epidemic more complex and challenging to control. This study offers vital insights into the genetic evolutionary branches and recent antigenic drift, providing crucial clues for updating the H7N9 vaccine seed virus and for disease prevention and control.
Dr. Liu is a postgraduate student at South China Agricultural University in Guangzhou, China. Her research interests are the epidemiology, pathogenesis, and control of major poultry diseases and zoonoses. Dr. Chen is a veterinarian at Guangzhou Animal Health Inspection Institute in Guangzhou, China. His research interests are animal disease prevention and control.
Acknowledgments
We appreciate the support of Guangzhou Animal Health Supervision Institute for our sampling work. We also gratefully acknowledge all data contributors, i.e., the Authors and their Originating laboratories responsible for obtaining the specimens, and their Submitting laboratories for generating the genetic sequence and metadata and sharing via the GISAID Initiative, on which this research is based.
In this study, we uploaded the hemagglutinin gene sequences of 23 H7N9 viruses isolates from 2021 to 2023. All sequencing data are available in the GISAID database (https://www.gisaid.org). (Accession number information is in Appendix Table 6.)
This work was supported by the Science and Technology Program of Guangdong Province (grant nos. 2022B1111010004 and 2021B1212030015), the China National Natural Science Foundation (grant no. 31972709), China Agriculture Research System of Ministry of Finance and Ministry of Agriculture and Rural Affairs (project no. CARS-41), China National Animal Disease Surveillance and Epidemiological Survey Program (2021–2025) (grant no. 202111), the Basic and Applied Basic Research Project of Guangzhou Basic Research Plan (grant no. 202102080576), and Guangdong Aib Polytechnic College Program (grant no. XJYB202105).
References
- Wang X, Jiang H, Wu P, Uyeki TM, Feng L, Lai S, et al. Epidemiology of avian influenza A H7N9 virus in human beings across five epidemics in mainland China, 2013-17: an epidemiological study of laboratory-confirmed case series. Lancet Infect Dis. 2017;17:822–32. DOIPubMedGoogle Scholar
- Ke C, Mok CKP, Zhu W, Zhou H, He J, Guan W, et al. Human infection with highly pathogenic avian influenza A(H7N9) virus, China. Emerg Infect Dis. 2017;23:1332–40. DOIPubMedGoogle Scholar
- Yang L, Zhu W, Li X, Chen M, Wu J, Yu P, et al. Genesis and spread of newly emerged highly pathogenic H7N9 avian viruses in mainland China. J Virol. 2017;91:e01277–17. DOIPubMedGoogle Scholar
- Senne DA, Panigrahy B, Kawaoka Y, Pearson JE, Süss J, Lipkind M, et al. Survey of the hemagglutinin (HA) cleavage site sequence of H5 and H7 avian influenza viruses: amino acid sequence at the HA cleavage site as a marker of pathogenicity potential. Avian Dis. 1996;40:425–37. DOIPubMedGoogle Scholar
- Qi W, Jia W, Liu D, Li J, Bi Y, Xie S, et al. Emergence and adaptation of a novel highly pathogenic H7N9 influenza virus in birds and humans from a 2013 human-infecting low-pathogenic ancestor. J Virol. 2018;92:e00921–17. DOIPubMedGoogle Scholar
- Gu J, Yan Y, Zeng Z, Wang W, Gao R, Hu J, et al. Characterization of two chicken origin highly pathogenic H7N9 viruses isolated in northern China. Vet Microbiol. 2022;268:
109394 . DOIPubMedGoogle Scholar - Byrne AMP, Reid SM, Seekings AH, Núñez A, Obeso Prieto AB, Ridout S, et al. H7N7 avian influenza virus mutation from low to high pathogenicity on a layer chicken farm in the UK. Viruses. 2021;13:259. DOIPubMedGoogle Scholar
- Yin X, Deng G, Zeng X, Cui P, Hou Y, Liu Y, et al. Genetic and biological properties of H7N9 avian influenza viruses detected after application of the H7N9 poultry vaccine in China. PLoS Pathog. 2021;17:
e1009561 . DOIPubMedGoogle Scholar - Wu Y, Hu J, Jin X, Li X, Wang J, Zhang M, et al. Accelerated evolution of H7N9 subtype influenza virus under vaccination pressure. Virol Sin. 2021;36:1124–32. DOIPubMedGoogle Scholar
- Chen J, Liu Z, Li K, Li X, Xu L, Zhang M, et al. Emergence of novel avian origin H7N9 viruses after introduction of H7-Re3 and rLN79 vaccine strains to China. Transbound Emerg Dis. 2022;69:213–20. DOIPubMedGoogle Scholar
- Jiang W, Hou G, Li J, Peng C, Wang S, Liu S, et al. Antigenic variant of highly pathogenic avian influenza A(H7N9) virus, China, 2019. Emerg Infect Dis. 2020;26:379–80. DOIPubMedGoogle Scholar
- Khare S, Gurry C, Freitas L, Schultz MB, Bach G, Diallo A, et al. GISAID’s role in pandemic response. China CDC Wkly. 2021;3:1049–51. DOIPubMedGoogle Scholar
- Zhang F, Bi Y, Wang J, Wong G, Shi W, Hu F, et al. Human infections with recently-emerging highly pathogenic H7N9 avian influenza virus in China. J Infect. 2017;75:71–5. DOIPubMedGoogle Scholar
- Zhang J, Ye H, Li H, Ma K, Qiu W, Chen Y, et al. Evolution and antigenic drift of influenza A(H7N9) viruses, China, 2017–2019. Emerg Infect Dis. 2020;26:1906–11. DOIPubMedGoogle Scholar
- Wu J, Ke C, Lau EHY, Song Y, Cheng KL, Zou L, et al. Influenza H5/H7 virus vaccination in poultry and reduction of zoonotic infections, Guangdong Province, China, 2017–18. Emerg Infect Dis. 2019;25:116–8. DOIPubMedGoogle Scholar
Figures
Table
Cite This ArticleOriginal Publication Date: April 19, 2024
1These first authors contributed equally to this article.
Table of Contents – Volume 30, Number 6—June 2024
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Weixin Jia or Ming Liao, College of Veterinary Medicine, South China Agricultural University, 483 Wushan Rd, Tianhe, Guangzhou, Guangdong 510642, China
Top