Volume 30, Number 8—August 2024
Online Report
Proposal for a Global Classification and Nomenclature System for A/H9 Influenza Viruses
Abstract
Influenza A/H9 viruses circulate worldwide in wild and domestic avian species, continuing to evolve and posing a zoonotic risk. A substantial increase in human infections with A/H9N2 subtype avian influenza viruses (AIVs) and the emergence of novel reassortants carrying A/H9N2-origin internal genes has occurred in recent years. Different names have been used to describe the circulating and emerging A/H9 lineages. To address this issue, an international group of experts from animal and public health laboratories, endorsed by the WOAH/FAO Network of Expertise on Animal Influenza, has created a practical lineage classification and nomenclature system based on the analysis of 10,638 hemagglutinin sequences from A/H9 AIVs sampled worldwide. This system incorporates phylogenetic relationships and epidemiologic characteristics designed to trace emerging and circulating lineages and clades. To aid in lineage and clade assignment, an online tool has been created. This proposed classification enables rapid comprehension of the global spread and evolution of A/H9 AIVs.
Avian influenza viruses (AIVs), including type A of the H9 subtype (A/H9), have a worldwide distribution in wild birds and are endemic in poultry populations in several countries in Asia, the Middle East, and Africa. Despite being classified as low pathogenicity, AIVs, strains of the A/H9N2 subtype are becoming an increased threat to domestic birds and humans (1,2). A/H9N2 subtype viruses can cause major economic damage to the poultry industry because of decreased egg production and increased mortality, related to co-infection with bacteria and other viruses (3–6). The economic consequences from this subtype have prompted several countries to adopt poultry vaccination policies to try to control the virus spread (7,8). Those policies have further driven genetic diversification of A/H9 AIVs (8–11).
Since 1998, viruses of the A/H9 subtype have occasionally crossed over into mammal species, leading to detection in humans, pigs, dogs, horses, pikas, minks, and bats (12). As of December 1, 2023, A/H9N2 was associated with 128 human infections, 90% of which were reported in China (13), and more than half of cases (52%) were identified during 2020–2023. Of note, this number may be underestimated because A/H9N2 infected humans do not often show obvious symptoms (14). Furthermore, A/H9N2 AIVs were identified as a source of internal gene mutations, leading to the emergence of several reassortants in human infection reports, such as H3N8, H5N6, H7N9, and H10N8 (15–21).
To date, different names have been adopted to describe A/H9 influenza virus lineages or sublineages and clades based on the hemagglutinin (HA) gene, often with differing names for the same group (5,22–27). The most widely accepted 4 primary lineages have multiple and complex names: the BJ/94 lineage is also known as Y280-like, G9-like, or h9.4.2 (represented by A/chicken/Beijing/1/94, A/chicken/Hong Kong/G9/97, or A/duck/Hong Kong/Y280/97); the G1 lineage is also known as h9.4.1 (represented by A/quail/Hong Kong/G1/97); the Y439 lineage is also known as Korean or h9.3 (represented by A/chicken/Korea/38349-p96323/96 or A/duck/Hong Kong/Y439/97); and the American lineage is also known as h9.1-h9.2 (represented by A/turkey/Wisconsin/1/1966 or A/turkey/Minnesota/511/1978). Additional sublineages and clades continue to emerge because of the rapid evolution of the A/H9 virus (8,22,23,26–29). The difference in clade classification and naming hinders comparison between studies and challenges critical discussion on the epidemiology, evolution, and spread of this subtype.
To address this issue, an international consortium of scientists from 22 laboratories in Europe, Asia, Africa, and the Americas was established to create a uniform system for classifying A/H9 viruses. This group was endorsed by the WOAH/FAO (World Organisation for Animal Health/Food and Agriculture Organization of the United Nations) Network of Expertise on Animal Influenza. By reconstructing a comprehensive phylogenetic history of all globally collected and publicly available HA sequences of A/H9 viruses, of any neuraminidase subtype, the consortium worked to create a harmonized classification and nomenclature system for A/H9 to be used in future epidemiologic and evolutionary studies.
Sequence Data and Metadata Preparation
To provide a comprehensive picture of A/H9 genetic diversity, we generated a dataset of the HA gene that included every H9-HA sequence available on the GISAID (https://www.gisaid.org) and GenBank public databases, which we accessed on July 18, 2022. We aligned the sequence dataset by using MAFFT v7.0 (30) and curated as described (Appendix 1). A final dataset containing 10,638 HA sequences and the related information, including accession numbers, resulted after our quality check process (Appendix 2 Table 1).
Phylogenetic Characterization
We generated a maximum-likelihood (ML) phylogenetic tree from the final complete dataset (n = 10,638) of the A/H9 virus HA genes by using IQ-TREE v1.6 (31) and applying the best-fitted nucleotide substitution model selected by ModelFinder (32). We performed clustering of the ML phylogenies of the complete dataset by using the pathogen-agnostic clustering tool PhyCLIP (Appendix 1 Figure 1) (33).
We conducted validation of lineages and clades identified with PhyCLIP by using multiple datasets and software to perform phylogenetic analyses. We assessed the robustness of our inference on a down-sampled dataset of 1,000 sequences, and then we assessed the nodal supports on a down-sampled dataset of 2,000 sequences. We selected sequences on the basis of phylogenetic diversity and calculated them as the sum of branch lengths of the minimal subtree spanned by a set of taxa included in the phylogenetic tree. We conducted this selection by using the phylogenetic diversity analyzer tool (34), starting from the ML tree obtained from the complete dataset. We generated ML trees from the 2 down-sampled datasets in IQ-TREE v2.1.3 by performing ultrafast-bootstrap resampling analysis (1,000 replications) (31,35). We used the best-fitted nucleotide substitution model selected by ModelFinder, implemented in IQ-TREE (32). We analyzed lineages separately to characterize the different clades within each lineage. We generated 3 datasets: Y_dataset, G_dataset, and B_dataset.
To assess the consistency of the clades, we conducted phylogenetic analyses for each dataset by using IQ-TREE v 2.1.3 (31) and PhyML v3.0 (36). We used ModelFinder, implemented in IQ-TREE (32), to select the best-fit nucleotide substitution model for each dataset. We assessed nodal supports in the IQ-TREE and PhyML analyses by using ultrafast-bootstrap and Shimodaira-Hasegawa (SH)–like branch supports (35,36).
To visualize the phylogenetic trees, we used FigTree version 1.4.2 (Figtree, http://tree.bio.ed.ac.uk/software/figtree). We calculated within and between clades average pairwise nucleotide distances (APD) by using MEGA X (p-distance) (37). We used TreeTime (https://treetime.readthedocs.io/en/latest) to reconstruct the ancestral sequence, which enabled us to infer the nonsynonymous nucleotide mutations leading to amino acid variations at each internal node (38).
Pilot Dataset
After the clade identification process, we created subsets for each lineage. The subsets are pilot_Y (86 sequences), pilot_G (105 sequences), and pilot_B (101 sequences); pilot_complete included all 3 lineages (292 sequences) (Appendix 3). We created the 4 subsets by using the phylogenetic diversity analyzer tool (34) and then manually selected sequences of underrepresented lineages and clades (Appendix 3). We implemented ML phylogenetic analyses with 1,000 ultrafastbootstrap and 1,000 nonparametric bootstrap replicates to ensure that the topology inferred from the larger datasets would be maintained with fewer sequences.
Online Tool Development
We created our online tool to automate the lineage classification of A/H9 influenza viruses. For the tool creation, we used Python (Python, https://www.python.org) and added an interactive display through the Django framework (Django, https://www.djangoproject.com). The tool assesses user-provided sequences to determine if they belong to the A/H9 subtype. If confirmed, the tool further classifies the sequences into their specific lineage and clade. The query sequences will be included in a pilot dataset, and a ML phylogenetic tree will be constructed by using IQ-TREE. The output from the tool includes the clade annotation and ML phylogenetic tree.
Principles of the Nomenclature System
The genomic characterization of A/H9 is needed to support the surveillance, prevention, and risk assessment of this globally prevalent subtype. A unified criterion for A/H9 genotyping is necessary to compare molecular epidemiology data obtained from different laboratories. We propose a workable and practical lineage classification and nomenclature for A/H9 AIVs on the basis of a comprehensive phylogenetic analysis of all available HA gene sequences. Our proposed nomenclature of A/H9 AIVs meets multiple requirements by capturing local and global patterns of virus genetic diversity in a timely and coherent manner, being robust and flexible enough to accommodate emerging virus diversity, providing support to track emerging clades when they exhibit amino acid or biologic and epidemiologic changes, and being informative and easily traceable while enabling the incorporation of the major and minor clades over time.
Clade Classification of A/H9 Viruses
We used a curated dataset of 10,638 global A/H9 HA sequences (Appendix 2 Table 1) to assess the phylogenetic relationships among globally circulating A/H9 viruses and establish a unified classification system for the virus. We applied 5 steps to assign and identify clades (Appendix 1 Figure 2) by integrating phylogenetic topology, branch support, genetic distances, epidemiologic information, and amino acid variants shared within each group. We defined clades on the basis of the following criteria: clades are assigned to monophyletic groups including >3 sequences obtained from viruses sampled over a period >3 years, indicating sustained transmission of the clade in >2 influenza seasons; clades have an ultrafast-bootstrap value >80% for the clade-defining node; HA proteins within the clade must share >1 amino acid mutation; an APD of >6% between each clade is recommended. However, to meet all other criteria, the APD may be slightly lower for certain clades.
Figure 1
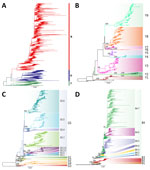
Figure 1. Phylogenetic trees and assigned clades of as part of a proposed global classification and nomenclature system for A/H9 influenza viruses. A) Maximum-likelihood (ML) phylogenetic tree obtained using the complete dataset....
Using those criteria, we defined 3 lineages, 18 first-order clades, 16 second-order clades, and 2 third-order clades (Figure 1). Nodal supports for each clade obtained from different analysis methods of each complete or down-sampled datasets are available (Appendix 1 Table 1). Each clade contains >1 amino acid change at the internal nodes (Appendix 1 Table 2). We found many mutations associated with host tropism, virulence, or antigenicity of the influenza virus (8), indicating that A/H9 AIVs of these clades may have undergone changes in biologic characteristics.
Nomenclature Criteria of A/H9 Lineages and Clades
To maintain consistency with traditional and commonly used lineage names for A/H9 (Y439, American, G1, and BJ/94 lineages) that convey information on spatial and biologic characteristics, we opted to label the 3 lineages as Y (to correspond with Y439 and American lineages), G (to correspond with G1 lineages), and B (to correspond with BJ/94 lineages). The numeric values, such as Y1 or G1 for the first-order clade, Y1.1 or G1.1 for the second-order clade, Y1.1.1 or G1.1.1 for the third-order clade, are used to identify clades that descended from the Y, G, and B lineages. In addition, viruses are classified as Y-like, G-like, or B-like if they belong to 1 of the identified lineages but do not fall within any clade. We provided a comparison between our proposed nomenclature system and some of the previously used A/H9 nomenclatures that enables rapid mapping of our lineages and clades to the name of the previously used lineages and clades (Appendix 1 Table 3).
Pilot Datasets of Lineages and Clades
Figure 2

Figure 2. Pilot maximum-likelihood phylogenetic tree of the A/H9 influenza virus gene sequences obtained by using the representative dataset (Appendix 3) for the Y lineage provided as part of a...
Figure 3

Figure 3. Pilot maximum-likelihood phylogenetic tree of the A/H9 influenza virus gene sequences obtained by using the representative dataset (Appendix 3) for the G lineage provided as part of a...
Figure 4

Figure 4. Pilot maximum-likelihood phylogenetic tree of the A/H9 influenza virus gene sequences obtained by using the representative dataset (Appendix 3) for the B lineage provided as part of a...
We generated 3 small datasets of ≈100 sequences containing representative viruses for each clade of the Y, G, and B lineages. To ensure that topology is maintained with a lower number of sequences, we performed ML phylogenetic reconstructions. All lineage and clade-defining branches are strongly supported (Appendix 1 Table 1) and the topologies of the generated pilot trees (Figure 2; Figure 3; Figure 4; Appendix 1 Figure 4) are consistent with the trees generated from larger datasets. All pilot datasets that contain sequences labeled according to lineage and clade are available (Appendix 3). Those pilot datasets can be used for rapid classification of newly sequenced A/H9 viruses according to our proposed classification scheme. If the lineage is unknown, the nucleotide A/H9 HA gene sequence or sequences under investigation can be analyzed by using the complete pilot dataset that includes all 3 lineages (Appendix 3). The complete pilot dataset will identify both the lineage and the specific clade according to clustering. If the lineage is known, the phylogenetic analysis can be performed by using the small and easily manageable pilot dataset specific for the lineage of interest, either pilot G, pilot Y, or pilot B (Appendix 3). The lineage and clade of an H9 sequence can also be identified by using the online tool.
Genetic and Epidemiologic Characteristics of Individual A/H9 Lineages and Clades
To determine whether the nomenclature system can reflect the real epidemiologic characteristics of A/H9 influenza viruses, we compiled a detailed description for those lineages and clades (Appendix 1 Tables 4–6; Appendix 2 Table 2) that includes the temporal, spatial, and host distribution. However, considering the limited number of sequences available for certain countries and the limited sampling data from wild birds, sampling bias cannot be excluded.
Y Lineage
Figure 5

Figure 5. Temporal distribution of each lineage and clade for A/H9 influenza viruses as part of a proposed global classification and nomenclature system for A/H9 influenza viruses. The heat map displays the...
Figure 6

Figure 6. Geographic distribution of each lineage and clade for A/H9 influenza viruses as part of a proposed global classification and nomenclature system for A/H9 influenza viruses. The heat map displays the...
The Y lineage (n = 622), including the previously named Y439 (or Korean, h9.3) and American (or h9.1–h9.2) lineages (27,39), was first identified in 1966 and is the oldest and most widespread lineage of A/H9 influenza viruses (40). In our dataset, the group contains sequences identified after 1976 because we removed earlier strains such as A/turkey/Wisconsin/1/1966 (Figure 5; Appendix 1 Table 4). The Y lineage has spread to every continent (Figure 6; Appendix 1 Table 4) and has evolved into 9 first-order (Y1–Y9) and 2 second-order (Y2.1 and Y2.2) well-supported clades (ultrafast-bootstrap >84, SH-like >0.85) (Figure 1, panel B). Clades of the Y lineage exhibited a high between-clade APD ranging from 7.10% to 18.30% (Appendix 1 Figure 3; Appendix 2 Table 2). Clades Y2.1 (South America, 2007–2017) and Y2.2 (United States, 1993–1996) showed the highest genetic distance from all other clades (15.37%–18.30%). Within-clade APD was <6% for all Y lineage clades, ranging from clade Y5 at 1.19% (United States, 2005–2007) to clade Y6 at 5.71% (Cambodia, Vietnam, 2009–2018).
Figure 7
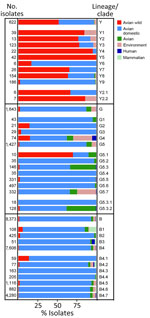
Figure 7. Host distribution of each lineage and clade for A/H9 influenza viruses as part of a proposed global classification and nomenclature system for A/H9 influenza viruses. The bar chart illustrates the...
Clades Y3, Y4, Y7, and Y8 were detected in >3 countries on >2 different continents, and clade Y8 has the widest geographic distribution: Australia, Asia, Europe, Africa, and North America. The remaining clades were identified in fewer countries. Clades Y1, Y2, and Y5 are exclusively from the Americas, and clade Y6 is specific to Southeast Asia (Cambodia and Vietnam). Detections of Y lineage viruses are equally distributed between wild and domestic birds, and 1 detection occurred in a mammal host (swine). The proportion of wild and domestic hosts varies across the identified clades. Domestic poultry accounts for 83% of clade Y6 detections and 96% of clade Y9 detections, whereas the other clades have been identified from mostly wild birds (64%–100%) (Figure 7; Appendix 1 Table 4).
G Lineage
The G lineage (n = 1643), previously known as G1 or h9.4.1 (27,39), has been circulating since 1997 (Figure 5; Appendix 1 Table 5). Domestic birds account for 85% of G lineage sequences (Figure 7), and the lineage is found in countries in Asia (61%) and Africa (38%) (Figure 6; Appendix 1 Table 5). According to the proposed clade classification criteria, we distinguished 5 first-order clades (G1–G5). Between-clade APD of the G lineage ranges from 4.81% to 13.59% (Appendix 1 Figure 3; Appendix 2 Table 2). Clade G5 shows the highest genetic distance from the other clades (10.94%–13.59%). Clade G5 was detected in 29 countries and was further divided into second-order (G5.1–G5.7) and third-order (G5.3.1–G5.3.2) subclades (Figure 1, panel C). The genetic distance between G1 and G2 is 5.81% and between G5.1 and G5.2 is 4.81%; those distances are less than the established 6% cutoff but are considered acceptable because they do not share a stable common root node. Within-clade APD was <6% for all clades, ranging from clade G1 (Israel, Jordan, Lebanon, 2003–2007) at 1.30% to clade G5.7 at 5.84% (Bangladesh, India, Kuwait, Pakistan, 2003–2022) (Appendix 1 Figure 3; Appendix 2 Table 2). All the identified clades are well supported (ultrafast-bootstrap >95; SH-like >0.73).
Most of the G clade viruses originate from domestic poultry, except for G5.1, which was found in 70% of the viruses sampled from wild avian species (Figure 7; Appendix 1 Table 5). Since 1999, human infections of viruses belonging to clades G4 (n = 4, 1999–2009), G5.3.2 (n = 1, 2015), G5.5 (n = 2, 2019), and G5.7 (n = 2, 2011–2019) have been detected.
B Lineage
The B lineage (n = 8,373), previously known as BJ/94, Y280, G9, or h9.4.2 (27,39), has been circulating since 1994 in domestic birds (92% of the sequences) from Asia, and >93% of the sequenced viruses originated from China (Figure 5; Figure 6; Figure 7; Appendix 1 Table 6). We divided lineage B into 11 well supported (ultrafast-bootstrap >81; SH-like >0.9) clades, consisting of 4 first-order clades (B1–B4) and 7 second-order clades (B4.1–B4.7) (Figure 1, panel D). Compared with the other lineages, clades of the B lineage exhibited lower between-clade APD, ranging from 4.48% to 12.06% (Appendix 1 Figure 3; Appendix 2 Table 2). Clade B4 shows the highest genetic distance from all the other clades (10.54%–12.06%). Clade B4 has had a considerable geographic expansion in the past 10 years. This clade affects 11 distinct countries in Asia and has further diversified into 7 second-order clades (B4.1–B4.7). As with the G lineage, the B lineage displayed a genetic distance between some clades of <6%. Within-clade APD was <6% for all clades, ranging from clade B4.4 at 2.18% (China, 2009–2020) to clade B4.7 at 5.04% (Cambodia, China, Japan, Laos, Myanmar, Russian Federation, Tajikistan, Vietnam, 2012–2021) (Figure 6; Appendix 1 Table 6; Appendix 2 Table 2).
Within the B lineage, all viruses sequenced in the past 5 years fall into clades B4.4–B4.7. Most of the viruses in each B clade were identified in poultry (Figure 7; Appendix 1 Table 6), and almost all clades contain viruses collected from mammal species. Most viruses recovered from human infections (82.9%, 34/41) fall within clade B4.7 (2014–2021).
Online Tool for Automatic Sequence Classification
To enhance the accessibility of our A/H9 influenza virus clade classification and nomenclature system, we developed an intuitive online tool (https://nmdc.cn/influvar/tools/H9aiv) with the aim to provide a user-friendly interface, making it easier for researchers and stakeholders to use and navigate the classification system. Users will be able to access the website and submit their A/H9 sequence file in the appropriate format. The tool will assign the corresponding clade and will provide a phylogenetic tree.
Establishing a unified nomenclature system for A/H9 influenza viruses is essential for the provision of communication to address the increasing global threat of the viruses. This system is characterized by 4 key hallmarks: comprehensiveness, robustness, inclusiveness, and practicality.
For comprehensiveness, we have created a system based on the HA gene sequence data of all the A/H9 viruses (including different neuraminidase subtypes) collected worldwide during 1976–2022. Those data enable understanding of the complete evolution of A/H9 AIVs on a regional and global scale, as well as the pattern of geographic and host distribution of lineages and clades. For example, the G and Y lineages are found in multiple continents, indicating a wide geographic diffusion, whereas the B lineage is isolated primarily in Asia and has occasional spillover to Europe through Russia. In addition, we present a record of all A/H9 AIVs clades, past and presently circulating, to provide a comprehensive understanding of the virus evolution trajectory in different host and geographic contexts.
Our system demonstrates robustness by being built from phylogenetic analysis of high-quality, nearly complete (>75%) HA gene sequences from multiple datasets (complete and down-sampled) and software packages. We have included all unique publicly available sequences, from a wide variety of origins and collection dates, to ensure the comprehensiveness and representativeness of our dataset. We only accepted high-supported clades consistently recognized across all trees, regardless of the datasets and approaches used, and characterized by a specific set of shared amino acid mutations. For example, there are 4 widely and internationally recognized lineages of A/H9 viruses, American, Y439, G1, and BJ/94 (Y280). However, because our classification approach revealed that the HA sequences of American-like and Y439-like lineages are phylogenetically closely related and share an ancestral node, they were combined into a single lineage and renamed as the Y lineage.
The inclusiveness of our system stems from the establishment of multiple criteria to identify and classify novel clades that emerge, incorporating phylogenetic evidence, molecular evidence, and epidemiologic features. Accommodating all the criteria can be challenging, because natural virus evolution is unlikely to always produce discrete boundaries. Among the criteria, the phylogenetic relationship and epidemiologic characteristics are the most useful for classification, whereas a certain degree of flexibility can be acceptable for bootstrap or APD value, provided there is strong evidence that the monophyletic clade exists and is epidemiologically relevant. This flexibility should enable the monitoring of ongoing epidemics in that clade, especially the emergence and evolution of antigenically distinct strains. The balance between the fixed criteria and topology or epidemiologic characters is also applied in the classification of H5 influenza viruses and SARS-CoV-2 (41,42).
Practicality is the most relevant hallmark of our system. We have fully considered the needs of users and have made major efforts to achieve full applicability of our system. Users can follow simple and clear criteria to identify novel clades, understand the biologic and epidemiologic significance of clades, and communicate results of their investigations through an informative nomenclature system. In assigning lineage names, we considered the traditional nomenclature in use to create consistency with historic classifications and to enable comparisons with previous studies.
In summary, our proposed nomenclature system and online tool for A/H9 AIVs is valuable for understanding the ongoing evolution and spread of this influenza subtype. Our nomenclature system will improve communication among international and local organizations and laboratories working on A/H9 viruses. The use of our system will enhance the global prevention and control capacity of A/H9 AIVs outbreaks.
Members of the International H9 Evolution Consortium: Celia Abolnik (South Africa), Ian Brown (United Kingdom), Charles Todd Davis (United States), Xiangjun Du (China), Shangang Jia (China), Erik A. Karlsson (Cambodia), Dong-Hun Lee (South Korea), Nicola S. Lewis (United Kingdom), Hualei Liu (China), Yudong Li (China), Yoshihiro Sakoda (Japan), Jianzhong Shi (China), David E Swayne (United States), Frank Y.K.Wong (Australia), and Jinfeng Zeng (China).
Dr. Fusaro is a scientist at the European Union Reference Laboratory for Avian Influenza and Newcastle Disease in Italy. Her primary research interests include molecular epidemiology, intra- and inter-host evolution, gene flow, and cross-species transmission of RNA viruses.
Acknowledgments
We thank the WOAH/FAO Network of Expertise on Animal Influenza for their technical expertise and leadership on this project. We thank Jie Cui, Xiang Li, and Francesca Ellero for their support and assistance.
Funding has been provided from the National Key Research and Development Program of China (grant nos. 2021YFD1800202 and 2022YFF0802403) and the National Natural Science Foundation of China (grant no. 32192450). Partial funding was provided by the EU through the NextGeneration EU-MUR PNRR Extended Partnership initiative on Emerging Infectious Diseases (project no. PE00000007, INF-ACT), the European Union’s Horizon 2020 Research and Innovation program (grant no. 874735), the Biological Sciences Research Council ecology and evolution of infectious diseases project (grant no. BB/V011286/1), Biological Sciences Research Council Institute Strategic Program Grant for the control of infectious diseases (grant no. BBS/E/D/20002173), and the Innovation and Technology Commission of the Hong Kong Special Administrative Region, China.
References
- Zhang J, Huang L, Liao M, Qi W. H9N2 avian influenza viruses: challenges and the way forward. Lancet Microbe. 2023;4:e70–1. DOIPubMedGoogle Scholar
- Li X, Shi J, Guo J, Deng G, Zhang Q, Wang J, et al. Genetics, receptor binding property, and transmissibility in mammals of naturally isolated H9N2 Avian Influenza viruses. PLoS Pathog. 2014;10:
e1004508 . DOIPubMedGoogle Scholar - Wang X, Liu K, Guo Y, Pei Y, Chen X, Lu X, et al. Emergence of a new designated clade 16 with significant antigenic drift in hemagglutinin gene of H9N2 subtype avian influenza virus in eastern China. Emerg Microbes Infect. 2023;12:
2249558 . DOIPubMedGoogle Scholar - Bonfante F, Mazzetto E, Zanardello C, Fortin A, Gobbo F, Maniero S, et al. A G1-lineage H9N2 virus with oviduct tropism causes chronic pathological changes in the infundibulum and a long-lasting drop in egg production. Vet Res (Faisalabad). 2018;49:83. DOIPubMedGoogle Scholar
- Pu J, Wang S, Yin Y, Zhang G, Carter RA, Wang J, et al. Evolution of the H9N2 influenza genotype that facilitated the genesis of the novel H7N9 virus. Proc Natl Acad Sci U S A. 2015;112:548–53. DOIPubMedGoogle Scholar
- Li C, Yu K, Tian G, Yu D, Liu L, Jing B, et al. Evolution of H9N2 influenza viruses from domestic poultry in Mainland China. Virology. 2005;340:70–83. DOIPubMedGoogle Scholar
- Dong J, Zhou Y, Pu J, Liu L. Status and challenges for vaccination against avian H9N2 influenza virus in China. Life (Basel). 2022;12:12. DOIPubMedGoogle Scholar
- Carnaccini S, Perez DR. H9 Influenza viruses: an emerging challenge. Cold Spring Harb Perspect Med. 2020;10:10. DOIPubMedGoogle Scholar
- Zhang N, Quan K, Chen Z, Hu Q, Nie M, Xu N, et al. The emergence of new antigen branches of H9N2 avian influenza virus in China due to antigenic drift on hemagglutinin through antibody escape at immunodominant sites. Emerg Microbes Infect. 2023;12:
2246582 . DOIPubMedGoogle Scholar - Yan W, Cui H, Engelsma M, Beerens N, van Oers MM, de Jong MCM, et al. Molecular and antigenic characterization of avian H9N2 viruses in southern China. Microbiol Spectr. 2022;10:
e0082221 . DOIPubMedGoogle Scholar - Pu J, Yin Y, Liu J, Wang X, Zhou Y, Wang Z, et al. Reassortment with dominant chicken H9N2 influenza virus contributed to the fifth H7N9 virus human epidemic. J Virol. 2021;95:95. DOIPubMedGoogle Scholar
- Peacock THP, James J, Sealy JE, Iqbal M. A global perspective on H9N2 avian influenza virus. Viruses. 2019;11:11. DOIPubMedGoogle Scholar
- Adlhoch C, Fusaro A, Gonzales JL, Kuiken T, Mirinavičiūtė G, Niqueux É, et al.; European Food Safety Authority; European Centre for Disease Prevention and Control; European Union Reference Laboratory for Avian Influenza. Avian influenza overview September-December 2023. EFSA J. 2023;21:
e8539 .PubMedGoogle Scholar - Chan RWY, Chan LLY, Mok CKP, Lai J, Tao KP, Obadan A, et al. Replication of H9 influenza viruses in the human ex vivo respiratory tract, and the influence of neuraminidase on virus release. Sci Rep. 2017;7:6208. DOIPubMedGoogle Scholar
- Yang R, Sun H, Gao F, Luo K, Huang Z, Tong Q, et al. Human infection of avian influenza A H3N8 virus and the viral origins: a descriptive study. Lancet Microbe. 2022;3:e824–34. DOIPubMedGoogle Scholar
- Wang Y, Niu S, Zhang B, Yang C, Zhou Z. The whole genome analysis for the first human infection with H10N3 influenza virus in China. J Infect. 2021:S0163–4453(21)00318-2.
- Bi Y, Chen Q, Wang Q, Chen J, Jin T, Wong G, et al. Genesis, evolution and prevalence of H5N6 avian influenza viruses in China. Cell Host Microbe. 2016;20:810–21. DOIPubMedGoogle Scholar
- Qi W, Zhou X, Shi W, Huang L, Xia W, Liu D, et al. Genesis of the novel human-infecting influenza A(H10N8) virus and potential genetic diversity of the virus in poultry, China. Euro Surveill. 2014;19:20841. DOIPubMedGoogle Scholar
- Liu D, Shi W, Gao GF. Poultry carrying H9N2 act as incubators for novel human avian influenza viruses. Lancet. 2014;383:869. DOIPubMedGoogle Scholar
- Liu D, Shi W, Shi Y, Wang D, Xiao H, Li W, et al. Origin and diversity of novel avian influenza A H7N9 viruses causing human infection: phylogenetic, structural, and coalescent analyses. Lancet. 2013;381:1926–32. DOIPubMedGoogle Scholar
- Liu WJ, Wu Y, Bi Y, Shi W, Wang D, Shi Y, et al. Emerging HxNy influenza A viruses. Cold Spring Harb Perspect Med. 2022;12:12. DOIPubMedGoogle Scholar
- Zhuang Q, Wang S, Liu S, Hou G, Li J, Jiang W, et al. Diversity and distribution of type A influenza viruses: an updated panorama analysis based on protein sequences. Virol J. 2019;16:85. DOIPubMedGoogle Scholar
- Li C, Wang S, Bing G, Carter RA, Wang Z, Wang J, et al. Genetic evolution of influenza H9N2 viruses isolated from various hosts in China from 1994 to 2013. Emerg Microbes Infect. 2017;6:
e106 . DOIPubMedGoogle Scholar - Shepard SS, Davis CT, Bahl J, Rivailler P, York IA, Donis RO. LABEL: fast and accurate lineage assignment with assessment of H5N1 and H9N2 influenza A hemagglutinins. PLoS One. 2014;9:
e86921 . DOIPubMedGoogle Scholar - Jiang W, Liu S, Hou G, Li J, Zhuang Q, Wang S, et al. Chinese and global distribution of H9 subtype avian influenza viruses. PLoS One. 2012;7:
e52671 . DOIPubMedGoogle Scholar - Fusaro A, Monne I, Salviato A, Valastro V, Schivo A, Amarin NM, et al. Phylogeography and evolutionary history of reassortant H9N2 viruses with potential human health implications. J Virol. 2011;85:8413–21. DOIPubMedGoogle Scholar
- Liu S, Ji K, Chen J, Tai D, Jiang W, Hou G, et al. Panorama phylogenetic diversity and distribution of Type A influenza virus. PLoS One. 2009;4:
e5022 . DOIPubMedGoogle Scholar - Dalby AR, Iqbal M. A global phylogenetic analysis in order to determine the host species and geography dependent features present in the evolution of avian H9N2 influenza hemagglutinin. PeerJ. 2014;2:
e655 . DOIPubMedGoogle Scholar - Guan Y, Shortridge KF, Krauss S, Webster RG. Molecular characterization of H9N2 influenza viruses: were they the donors of the “internal” genes of H5N1 viruses in Hong Kong? Proc Natl Acad Sci U S A. 1999;96:9363–7. DOIPubMedGoogle Scholar
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80. DOIPubMedGoogle Scholar
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74. DOIPubMedGoogle Scholar
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587–9. DOIPubMedGoogle Scholar
- Han AX, Parker E, Scholer F, Maurer-Stroh S, Russell CA. Phylogenetic clustering by linear integer programming (PhyCLIP). Mol Biol Evol. 2019;36:1580–95. DOIPubMedGoogle Scholar
- Chernomor O, Minh BQ, Forest F, Klaere S, Ingram T, Henzinger M, et al. Split diversity in constrained conservation prioritization using integer linear programming. Methods Ecol Evol. 2015;6:83–91. DOIPubMedGoogle Scholar
- Minh BQ, Nguyen MAT, von Haeseler A. Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol. 2013;30:1188–95. DOIPubMedGoogle Scholar
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–21. DOIPubMedGoogle Scholar
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–9. DOIPubMedGoogle Scholar
- Sagulenko P, Puller V, Neher RA. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018;4:
vex042 . DOIPubMedGoogle Scholar - Guo YJ, Krauss S, Senne DA, Mo IP, Lo KS, Xiong XP, et al. Characterization of the pathogenicity of members of the newly established H9N2 influenza virus lineages in Asia. Virology. 2000;267:279–88. DOIPubMedGoogle Scholar
- Homme PJ, Easterday BC. Avian influenza virus infections. I. Characteristics of influenza A-turkey-Wisconsin-1966 virus. Avian Dis. 1970;14:66–74. DOIPubMedGoogle Scholar
- World Health Organization/World Organization for Animal Health/Food and Agriculture Organization H5N1 Evolution Working Group. Toward a unified nomenclature system for highly pathogenic avian influenza virus (H5N1). Emerg Infect Dis. 2008;14:
e1 . DOIGoogle Scholar - Rambaut A, Holmes EC, O’Toole Á, Hill V, McCrone JT, Ruis C, et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol. 2020;5:1403–7. DOIPubMedGoogle Scholar
Figures
Cite This ArticleOriginal Publication Date: July 16, 2024
1These first authors contributed equally to this article.
2Members of The International H9 Evolution Consortium are listed at the end of this article.
Table of Contents – Volume 30, Number 8—August 2024
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Jinhua Liu, China Agricultural University, No. 2 Yuanmingyuan West Rd, Beijing 100193, China
Top