Volume 30, Number 8—August 2024
Online Report
Proposal for a Global Classification and Nomenclature System for A/H9 Influenza Viruses
Figure 1
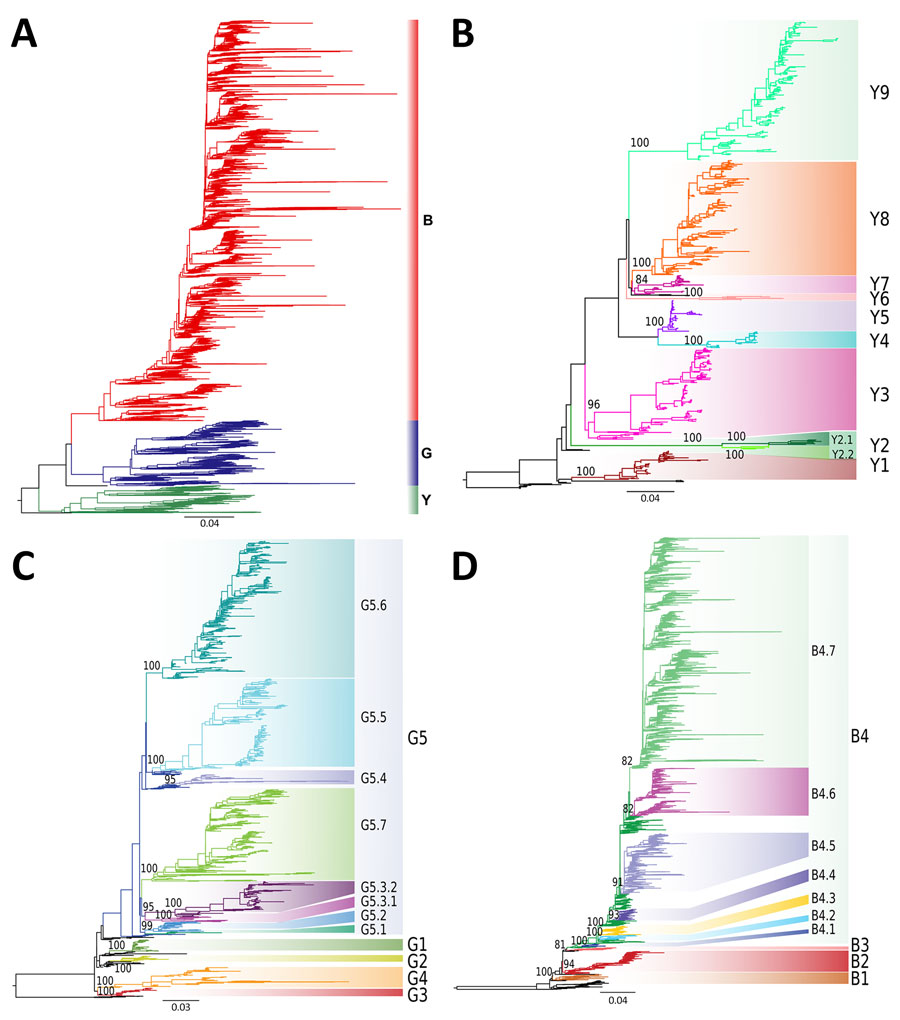
Figure 1. Phylogenetic trees and assigned clades of as part of a proposed global classification and nomenclature system for A/H9 influenza viruses. A) Maximum-likelihood (ML) phylogenetic tree obtained using the complete dataset. The branches are colored according to the 3 identified lineages. A/quail/Hong Kong/A28945/1988 and A/Quail/Hong Kong/AF157/1992 were used as an outgroup black). B) ML phylogenetic tree of the Y lineage dataset. Six sequences of the G lineage were used as the outgroup. Numbers next to the clade-defining nodes represent ultra-fast bootstrap supports. Clades are labeled and marked in colors. C) ML phylogenetic tree of the G lineage. Six sequences of the Y lineage were selected as the outgroup. Numbers next to the clade-defining nodes represent ultra-fast bootstrap supports. Clades are labeled and marked in colors. D) ML phylogenetic tree of the B lineage. Six sequences of the Y lineage were used as the outgroup. Numbers next to the clade-defining nodes represent ultra-fast bootstrap supports. Clades are labeled and marked in colors. Scale bars indicate substitutions per site.
1These first authors contributed equally to this article.
2Members of The International H9 Evolution Consortium are listed at the end of this article.