Volume 31, Supplement—May 2025
SUPPLEMENT ISSUE
Supplement
Respiratory Virus Detection and Sequencing from SARS-CoV-2–Negative Rapid Antigen Tests
Cite This Article
Citation for Media
Abstract
Genomic epidemiology offers insight into the transmission and evolution of respiratory viruses. We used metagenomic sequencing from negative SARS-CoV-2 rapid antigen tests to identify a wide range of respiratory viruses and generate full genome sequences. This process offers a streamlined mechanism for broad respiratory virus genomic surveillance.
The COVID-19 pandemic highlighted the importance of genomic epidemiology in understanding virus transmission and evolution, informing essential countermeasures from nonpharmaceutical interventions to vaccines. Massive global efforts in SARS-CoV-2 genomic surveillance were made possible by widespread diagnostic testing and the growth of new infrastructure and methods for sequencing and analysis (1). Most genomic surveillance pipelines in the United States obtained residual SARS-CoV-2–positive samples from clinical, public health, and commercial laboratories. That strategy was effective during the pandemic but difficult to maintain with the rise of at-home rapid antigen tests (2,3). As traditional sample sources declined, our group and others demonstrated that residual samples from rapid antigen tests could be used to generate and analyze full SARS-CoV-2 sequences for genomic surveillance (4–6).
In this study, we build upon that work by identifying, sequencing, and analyzing other respiratory viruses using residual swab samples from negative BinaxNOW COVID-19 antigen tests (Abbott, https://www.abbott.com). This multivirus approach is key because SARS-CoV-2 has transitioned to an endemic virus whose symptoms resemble those of other respiratory viruses (7). Thus, there is both a need for broad testing and an opportunity to expand genomic surveillance for respiratory viruses using self-collected samples.
In brief, participants were enrolled in a parent study evaluating novel viral diagnostic tests through the RADx program at the Atlanta Center for Microsystems Engineered Point-of-Care Technologies (Atlanta, GA, USA). The study protocol was approved by the Emory University Institutional Review Board and the Grady Health Research Oversight Committee (both in Atlanta). We performed RNA metagenomic sequencing as described (8), obtaining a median of 5.8 million reads per sample (Appendix 1; Appendix 2). We used a 3-step bioinformatic approach to detect viruses (Appendix 1 Figure 1) using KrakenUniq (https://github.com/fbreitwieser/krakenuniq), blastn (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome), and reference mapping (https://github.com/briannajeanne/metagen/tree/main). Our final criterion required coverage of >10% of the viral genome, or reads mapping to >3 nonoverlapping regions of the viral genome with >80% identity, similar to clinical diagnostic criteria that have previously been used for metagenomic sequencing (9).
We collected negative BinaxNOW test samples from 53 persons during April–August 2023 (Appendix 1 Table), a period during which 68% of the BinaxNOW tests in the parent study were negative. All persons were symptomatic at the time of testing (Table), and the median interval between symptom onset and testing was 2 (range 0–9) days. Reverse transcription PCR (RT-PCR) was positive for influenza B in 3 samples and negative for influenza A, respiratory syncytial virus, and SARS-CoV-2 in all samples (Appendix 2).
Figure 1
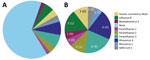
Figure 1. Frequency of human pathogenic respiratory viruses found in 53 residual samples from SARS-CoV-2–negative BinaxNOW tests (Abbott, https://www.abbott.com) in study of respiratory virus detection and sequencing from negative rapid...
Metagenomic sequencing identified a low level of SARS-CoV-2 in 1 sample and a different pathogenic human respiratory virus in 17 (33%) of the other 52 samples (Appendix 2). We detected parainfluenza viruses (n = 7), rhinoviruses (n = 5), influenza B (n = 3), seasonal coronaviruses (n = 2), and adenovirus (n = 1) (Figure 1). In 1 sample, we detected both influenza B and parainfluenza 2. In another sample positive for influenza B by RT-PCR, metagenomic sequencing did not identify influenza but identified human mastadenovirus E. Thus, excluding SARS-CoV-2, we detected a total of 18 viruses across 17 samples.
The duration of time between sample collection and nucleic acid extraction was similar for samples with a virus detected (median 6 [range 4–12] days) and samples with no virus detected (median 7 [range 5–19] days). RT-PCR for RNase P was positive in all samples tested, and the percentage of human reads was similar between samples with and without viruses detected (p = 0.07 by Mann-Whitney U test) (Appendix 2). We saw no difference in the total number of reads obtained for samples with and without viruses detected (p = 0.29 by Mann-Whitney U test).
We compared potential differences in symptoms between persons in whom a virus was detected and those in whom no virus was detected and observed the following disparities: congestion (83% vs. 63%), sore throat (78% vs. 54%), chills (61% vs. 37%), and headache (72% vs. 49%) (Table). However, none of those differences were statistically significant. The time between symptom onset and testing was similar between persons with a virus detected (median 2 [range 0–9] days) and those without a virus detected (median 2 [range 0–6] days).
Of the 18 viruses detected, we generated full viral genome sequences from 11 (61%) with >90% coverage and 71- to 24,000-fold depth (Appendix 2). Those 11 sequences consisted of parainfluenza 3 (4/4 samples), parainfluenza 2 (1/2), rhinovirus (5/5), and influenza B (1/3).
Figure 2
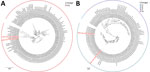
Figure 2. Phylogenetic analysis of parainfluenza 3 virus sequences in study of respiratory virus detection and sequencing from SARS-CoV-2–negative rapid antigen tests. The names of sequences obtained in this study are bold...
We performed phylogenetic analysis of parainfluenza 3 as a proof-of-concept for genomic epidemiology studies and found substantial diversity. Using the lineage classification system described in Lee et al. (10), 2 of our sequences clustered with lineage A1 sequences from 2019–2023 (Figure 2, panel A), another clustered with lineage C sequences from Japan in 2023, and the fourth with lineage C sequences from the United States collected during 2015–2017 (Figure 2, panel B), all with high bootstrap support (Appendix 1 Figure 2). Of note, only ≈450 complete parainfluenza 3 virus sequences are available; the data from our small study represent nearly 1% of this number, underscoring the opportunity to easily expand genomic surveillance using this approach.
Figure 3

Figure 3. Plot of the viral taxa (rows) that were detected in each sample (columns) in study of respiratory virus detection and sequencing from SARS-CoV-2–negative rapid antigen tests. A) Results from samples...
In addition to human pathogenic respiratory viruses, we detected >100 viruses of no clinical significance, including bacteriophages and plant viruses, many of which were also detected in our negative controls (Figure 3; Appendix 1 Figures 3, 4). Similarly, we found mastadenovirus C in about one third of all samples and negative controls, all with low genome coverage (Appendix 3). Those findings are all consistent with environmental or reagent contaminants. Herpesviruses were reported in many samples by KrakenUniq and blastn but generally were not confirmed by reference mapping. One adult participant had confirmed detection of human herpesvirus 6, which, given the participant’s age, more likely reflects latent virus than acute infection. Overall, 1,367 viral taxa were identified by KrakenUniq, only 254 (18.6%) were confirmed by blastn, and only 137 (53.9% [10% of total]) met our criteria for detection (Appendix 3), highlighting the importance of confirmatory steps in metagenomic analysis.
Our study demonstrates that RNA metagenomic sequencing of residual swab samples from negative BinaxNOW COVID-19 tests can be used to detect a broad range of respiratory viruses, including rhinoviruses, parainfluenza viruses, influenza B, seasonal coronaviruses, and adenovirus. All of those viruses have overlapping symptoms, both with one another and with SARS-CoV-2, underscoring the need for multivirus testing approaches. Although our study was not designed for clinical diagnosis, metagenomic sequencing is increasingly used clinically, and our results illustrate the need for rigorous analysis techniques and careful interpretation.
Of note, only 33% of samples had a human pathogenic respiratory virus. This finding is similar to that of our previous study, in which alternative respiratory viruses were detected in only 40% of SARS-CoV-2–negative persons using residual clinical samples early in the pandemic (8). Possible explanations include persons with a noninfectious syndrome, a bacterial or other nonviral infection, or a virus present at a low level. Some persons could also have been infected with a DNA virus not optimally captured by RNA sequencing. However, we detected adenovirus, the most prevalent respiratory DNA virus. Among common RNA viruses, we did not detect influenza A or respiratory syncytial virus, which we attribute to the winter-predominant seasonality of these viruses, whereas our samples were collected in spring and summer.
Of note, of the 18 viruses detected, we were able to generate full viral genome sequences from 11 (61%) using moderate sequencing depths. Thus, the single laboratory technique of metagenomic sequencing can not only identify diverse respiratory viruses but also contribute to their genomic surveillance. The surprisingly high depth of genome coverage achieved for many sequences indicates that throughput and cost can be improved by reducing total sequencing reads from each sample in future studies.
By combining metagenomic sequencing with the use of residual antigen test samples, we demonstrate a mechanism for convenient and broad respiratory virus surveillance. Our study used BinaxNOW tests, which conveniently preserve the used swab within the kit cassette; future work is needed to evaluate this approach using rapid antigen test strips themselves, as previously demonstrated for SARS-CoV-2 sequencing (5). In addition, future studies would benefit from a regulatory framework in which, after rigorous analysis and careful interpretation, clinically significant results can be returned to study participants, who are likely curious about the presence of other respiratory viruses when rapid antigen testing is negative for COVID-19. In conclusion, our study illustrates that residual samples from self-collected antigen tests can be a powerful sample source for investigating the genomic epidemiology of a broad range of respiratory viruses, building upon the strong foundations for viral surveillance established during the COVID-19 pandemic.
Ms. Jules is currently a research specialist in the Department of Pathology and Laboratory Medicine in the Emory University School of Medicine. She will be applying to medical school with the aspiration of becoming a family doctor and expanding healthcare to underserved communities.
Acknowledgments
We thank the study participants.
All raw sequencing data (cleaned of human reads) is available in the National Center for Biotechnology Information Sequence Read Archive under BioProject PRJNA1144955, and assembled virus genome sequences are available in GenBank with accession numbers listed in Appendix 2.
This work was supported by National Institutes of Health (NIH) awards U54 EB027690 02S1, U54 EB027690 03S1, and U54EB027690 03S2 UL1 TR002378 and by the Centers for Disease Control and Prevention–funded Georgia Pathogen Genomics Center of Excellence contract 40500-050-23234506. B.B. was supported by NIH award F31ES031845. This study was supported in part by the Emory Integrated Genomics Core (EIGC) (RRID:SCR_023529), which is subsidized by the Emory University School of Medicine and is one of the Emory Integrated Core Facilities. Additional support was provided by the Georgia Clinical & Translational Science Alliance of the NIH under award UL1TR002378. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the NIH.
References
- Oude Munnink BB, Worp N, Nieuwenhuijse DF, Sikkema RS, Haagmans B, Fouchier RAM, et al. The next phase of SARS-CoV-2 surveillance: real-time molecular epidemiology. Nat Med. 2021;27:1518–24. DOIPubMedGoogle Scholar
- Ritchey MD, Rosenblum HG, Del Guercio K, Humbard M, Santos S, Hall J, et al. COVID-19 self-test data: challenges and opportunities—United States, October 31, 2021–June 11, 2022. MMWR Morb Mortal Wkly Rep. 2022;71:1005–10. DOIPubMedGoogle Scholar
- Rader B, Gertz A, Iuliano AD, Gilmer M, Wronski L, Astley CM, et al. Use of at-home COVID-19 tests—United States, August 23, 2021–March 12, 2022. MMWR Morb Mortal Wkly Rep. 2022;71:489–94. DOIPubMedGoogle Scholar
- Nguyen PV, Carmola LR, Wang E, Bassit L, Rao A, Greenleaf M, et al. SARS-CoV-2 molecular testing and whole genome sequencing following RNA recovery from used BinaxNOW COVID-19 antigen self tests. J Clin Virol. 2023;162:
105426 . DOIPubMedGoogle Scholar - Martin GE, Taiaroa G, Taouk ML, Savic I, O’Keefe J, Quach R, et al. Maintaining genomic surveillance using whole-genome sequencing of SARS-CoV-2 from rapid antigen test devices. Lancet Infect Dis. 2022;22:1417–8. DOIPubMedGoogle Scholar
- Hassouneh SA, Trujillo A, Ali S, Cella E, Johnston C, DeRuff KC, et al. Antigen test swabs are comparable to nasopharyngeal swabs for sequencing of SARS-CoV-2. Sci Rep. 2023;13:11255. DOIPubMedGoogle Scholar
- Geismar C, Nguyen V, Fragaszy E, Shrotri M, Navaratnam AMD, Beale S, et al. Symptom profiles of community cases infected by influenza, RSV, rhinovirus, seasonal coronavirus, and SARS-CoV-2 variants of concern. Sci Rep. 2023;13:12511. DOIPubMedGoogle Scholar
- Babiker A, Bradley HL, Stittleburg VD, Ingersoll JM, Key A, Kraft CS, et al. Metagenomic sequencing to detect respiratory viruses in persons under investigation for COVID-19. J Clin Microbiol. 2020;59:e02142–20. DOIPubMedGoogle Scholar
- Miller S, Naccache SN, Samayoa E, Messacar K, Arevalo S, Federman S, et al. Laboratory validation of a clinical metagenomic sequencing assay for pathogen detection in cerebrospinal fluid. Genome Res. 2019;29:831–42. DOIPubMedGoogle Scholar
- Lee K, Park K, Sung H, Kim MN. Phylogenetic lineage dynamics of global parainfluenza virus type 3 post-COVID-19 pandemic. MSphere. 2024;9:
e0062423 . DOIPubMedGoogle Scholar
Figures
Table
Cite This Article1These first authors contributed equally to this article.
Table of Contents – Volume 31, Supplement—May 2025
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Anne Piantadosi, Woodruff Memorial Research Building, 101 Woodruff Cir, Atlanta, GA 30322, USA
Top