Volume 31, Number 6—June 2025
Synopsis
Genomic Surveillance of Climate-Amplified Cholera Outbreak, Malawi, 2022–2023
Cite This Article
Citation for Media
Abstract
In the aftermath of 2 extreme weather events in 2022, Malawi experienced a severe cholera outbreak; 59,325 cases and 1,774 deaths were reported by March 31, 2024. We generated 49 Vibrio cholerae full genomes from isolates collected during December 2022–March 2023. Phylogenetic and phylogeographic methods confirmed that the Malawi outbreak strains originated from Pakistan’s 2022 cholera outbreak. That finding aligns with substantial travel between the 2 countries. The estimated most recent ancestor of this lineage was from June–August 2022, coinciding with Pakistan’s floods and cholera surge. Our analysis indicates that major floods in Malawi contributed to the outbreak; reproduction numbers peaked in late December 2022. We conclude that extreme weather events and humanitarian crises in Malawi created conditions conducive to the spread of cholera, and population displacement likely contributed to transmission to susceptible populations in areas relatively unaffected by cholera for more than a decade.
Cholera is an acute diarrheal disease caused by ingestion of food or water contaminated with the bacterium Vibrio cholerae (1). Since mid-2021, the seventh cholera pandemic, associated with the V. cholerae O1 El Tor biotype, has been on an acute upsurge. Several large outbreaks have occurred in endemic and nonendemic countries; countries in Africa were particularly heavily affected (2). Those outbreaks have been driven by multiple factors, including extreme weather events, humanitarian crises, overlapping health emergencies (particularly during the COVID-19 pandemic), and consequently overstretched health systems (3,4).
In Malawi, cholera was first reported in 1973 and has been endemic since 1998. Annual increases in incidence occur during the rainy season (November–May, average temperature 30°C–35°C), particularly in the southern part of the country (5). During 2022–2024, Malawi experienced a widespread cholera outbreak that persisted throughout the dry season (May–November, average temperature 13°C); 59,325 cases and 1,774 deaths were reported as of March 31, 2024 (6). The outbreak in Malawi unfolded amidst a global surge in cholera outbreaks, intensifying the scarcity of vaccines, tests, and treatments (7). Several other countries in southeastern Africa, in particular Mozambique, South Africa, Tanzania, Zambia, and Zimbabwe, were experiencing outbreaks concomitant with the outbreak in Malawi (7).
Previous genomic analysis has revealed cholera epidemics in Africa to be associated with transcontinental transmission of V. cholerae O1 El Tor sublineages from Asia, followed by regional cross-border spread within Africa (8,9). To explore the origin and drivers of the 2022–2023 Malawi cholera outbreak, the Public Health Institute of Malawi (PHIM) partnered with the Centre for Epidemic Response and Innovation (CERI), a specialized genomics facility of the Africa Centres for Disease Control and Prevention and World Health Organization Regional Office for Africa (AFRO) (10), to perform in-country genomic sequencing of V. cholerae. Using phylogenetic and phylogeographic methods alongside epidemiologic modeling that incorporates flooding and vaccination data, we investigated the genomic epidemiology of the cholera outbreak in Malawi, while delving into its climate-amplified implications.
Ethics, Sample Selection, Culture, and DNA Extraction
To investigate the origin of the current cholera outbreak in Malawi, the Public Health Institute of Malawi, supported by CERI and the Climate Amplified Diseases and Epidemics (CLIMADE) program, performed onsite genomic sequencing of local isolates. Isolates were anonymized and did not contain any personal identifiers. The Stellenbosch University Health Research Ethics Committee approved the CLIMADE initiative (BES-2023-24266). The Public Health Institute of Malawi and Ministry of Health National Health Sciences Research Committee approved this study.
We collected fecal samples from patients with cholera symptoms who sought care in district and central hospitals. We used rapid diagnostic tests (RDTs) to identify the presence of V. cholerae and then performed culture on all samples that were RDT positive. We performed drug susceptibility testing on all positive cultures. We sent all positive culture plates to the National Genomic Sequencing Reference Laboratory for DNA extraction; we used the QIAamp DNA Mini Kit 51304 (QIAGEN, https://www.qiagen.com) for extraction and Qubit High Sensitivity DNA kit Q32854 (Thermo Fisher Scientific, https://www.thermofisher.com) on Qubit 4 for quantification. We obtained DNA extracts from 70 V. cholerae isolates from samples collected during December 2022–February 2023 from the Southern, Central, and Northern Regions of Malawi. We extracted demographic, clinical, and diagnostic data from the routine cholera surveillance system.
Sequencing
We prepared libraries using the Illumina DNA library preparation kit and Nextera CD indexes (Illumina, https://www.illumina.com), according to the manufacturer’s protocol. We performed whole-genome sequencing using the NextSeq 1000 instrument with P2 (300) cycle kit reagents (Illumina). We have deposited the genomic sequences into the National Center for Biotechnology Information Sequence Read Archive (BioProject no. PRJNA967700).
Assembly and High-Quality Single-Nucleotide Polymorphism Calling
We obtained full genomes and single-nucleotide polymorphism (SNP) alignments with our CholeraSeq pipeline (https://github.com/CERI-KRISP/CholeraSeq). In brief, we assessed read quality and trimmed all residual adaptors with fastp (11) and used Snippy version 4.6.0 (https://github.com/tseemann/snippy) to perform reference-based assembly against the N16961 strain (GenBank accession nos. NZ_CP028827.1, NZ_CP028828.1) before assembly. We set FreeBayes variant calling thresholds (E. Garrison, G. Marth, unpub. data, https://arxiv.org/abs/1207.3907) as >10× for site coverage, >60 for mapping quality, and >90% for base concordance. We merged individual vcf files using bctfools version 1.15 (12). We ran core genome alignment through fastBaps version 1.0.8 (13) before recombination screening with Gubbins version 3.2.1 (14). We manipulated FASTA files with seqkit version 2.0.0 (15) and the Biostrings R package version 2.58 (https://bioconductor.org/packages/release/bioc/html/Biostrings.html). We extracted parsimony informative sites from consensus genome alignments in MEGAX version 10.0.3 (16). We downloaded all available V. cholerae whole-genome sequencing experiments from the National Center for Biotechnology Information Short Read Archive and the European Nucleotide Archive. We assembled paired-end reads and called SNPs using the same methodology applied to the newly sequenced Malawi strains.
Phylogenetic Inference with Worldwide Cholera Dataset
We inferred a maximum-likelihood phylogenetic tree from the parsimony informative sites using IQ-TREE (17) to investigate the genetic relationship of the Malawi outbreak to that of other strains from around the world (sampled during 1957–2023; N = 2,778) (Appendix 1 Table 1). The reference set included 1,160 sequences from Africa, 898 from the Americas, 693 from Asia, 26 from Europe, and 1 from Oceania. We determined phylogenetic signal using a likelihood mapping test in IQ-TREE (17). We used Treetime (18) to obtain a maximum-likelihood tree scaled in time employing a standard mutation rate of 0.0179 substitutions/SNP site/year, as estimated by phylodynamic inference, after rerooting the tree by oldest tip.
Phylodynamic Inference
We investigated the phylogenetic relationships of the 49 new clinical strains from Malawi with strains within a monophyletic multicountry clade containing 31 strains collected in 2022 from Pakistan, 49 publicly available strains from the same outbreak in Malawi (19), 114 strains collected from South Africa in 2023 (20), and 20 strains collected from Zimbabwe in 2023 (21) (Appendix 1 Table 2). We determined phylogenetic signal using the likelihood mapping test in IQ-TREE (17) and estimated temporal signal by plotting the root-to-tip divergence using TempEst (22). We used the Bayesian framework to infer a posterior distribution of trees and estimate the time of the most recent common ancestor (tMRCA) of the sampled sequences. We considered different molecular clock models (strict or uncorrelated relaxed molecular clock) and demographic priors (constant or Bayesian Skygrid) (23). We used BEASTX version 10.5.0-β5 (24) to run Markov chain Monte Carlo samplers for 500 million generations, sampling every 50,000 generations, which was sufficient to achieve mixing of the Markov chain as evaluated by effective sampling size >200 for all parameter estimates under a given model. We performed hypothesis testing for best molecular clock, demographic model, by obtaining marginal likelihood estimates via path sampling and stepping-stone methods for each model to be compared, then calculating the Bayes factor (BF). BF is the ratio of the of the null (H0) and the alternative hypothesis (HA) marginal likelihood estimation s (25), where lnBF<0 indicates support for H0; lnBF<2, negligible difference; 2<lnBF<6, strong support for HA; and lnBF>6, decisive support for HA46 (Appendix 2 Table 1). We estimated the mutation rate at 0.0179 substitutions/SNP site/year, which is in line with previous rates for cholera (26,27). We obtained the maximum clade credibility (MCC) tree from the posterior distribution of trees using optimal burn-in with TreeAnnotator (https://beast.community/programs). We manipulated the MCC phylogeny in R using the package ggtree as described (28) for publishing purposes. We inferred the geographic origin of the epidemic in Malawi using discrete trait model with asymmetric transition (migration) and Bayesian stochastic search variable selection, enforcing the best molecular clock and demographic model (uncorrelated relaxed molecular clock and Bayesian Skygrid) in BEASTX. BF values of 3–20 indicate positive evidence, values of 20–150 indicate strong evidence, and values >150 indicate very strong evidence (29,30) (Appendix 2 Table 2). We investigated cholera movement across Malawi, South Africa, and Zimbabwe by ancestral state reconstruction using continuous traits with the migration extension of TreeTime (18). We used a custom Python script to estimate dates of exchange events. We mapped results using the R packages maptools, raster, rgdal, and sf (The R Project for Statistical Computing, https://www.r-project.org). XML, MCC tree, ML phylogeny, and Treetime migration files are available at https://github.com/cmavian/cholera_Malawi_2022-2023.
We used the WebPlotDigitizer tool (https://apps.automeris.io/wpd) to extract data from a WHO report on cholera in the Africa region (https://iris.who.int/bitstream/handle/10665/366745/AFRO%20Cholera%20Bulletin.06.pdf). The report provided daily information on cholera cases and deaths within Malawi until April 4, 2023.
Estimation of Time Varying Reproduction Number
We inferred the instantaneous reproduction number over time (Rt), at the national level, for Malawi using a semimechanistic Bayesian framework described in Bhatt et al. (31). To estimate the model, we used the epidemia package in R (https://imperialcollegelondon.github.io/epidemia), which enables us to estimate the effects of flooding and vaccinations as covariates. The estimate of Rt is based on daily national case counts, flooding, and vaccination data. We report the mean Rt estimate and 95% credible interval (CrI), a comparison of the observed and fitted case counts, and the effect sizes of our covariates (Appendix 2).
To check for robustness of our results, we estimated Rt with both flooding and vaccinations, only flooding, and with no covariates. We also confirmed that the estimation of Rt without any covariates is consistent with that of EpiEstim (32), which is an alternative implementation of a branching process model in R. Our Rt estimates were more stable because of the weekly random walk we used instead of daily data.
Flooding Data and Processing
We obtained real-time or near–real-time flooding data by remotely sensed Earth observation imagery (33). Three sources are the Sentinels in Europe (34), the US National Aeronautics and Space Administration LANCE MODIS NRT global flood product (MCDWD) product using images generated from the Moderate Resolution Imaging Spectroradiometer instrument (https://www.earthdata.nasa.gov/global-flood-product), and Flood version 1.0 (35) using images generated from the Visible Infrared Imaging Radiometer Suite instrument available at the RealEarth website (https://floods.ssec.wisc.edu) and associated archives (https://jpssflood.gmu.edu). Sentinels in Europe are generally used to provide data for emergency response rather than long-term archives; MCDWD provided a long-term time series through 2022; and Flood provides ongoing daily and 5-day composites, which were downloaded for all of 2022 and then until April 13, 2023. Image values distinguish normal open water (value <99) from flood water, for which values >100 represent percentage flooding plus 100. The 5-day composite is the maximum value recorded during the period. The files are in simple tiled geotiff format; 2 tiles are required to cover the whole of Malawi, which must be combined and cropped before analysis.
International Passenger Flight Data
We evaluated travel data generated from the International Air Transport Association (36) to quantify passenger volumes originating from international airports and arriving in Malawi. International Air Transport Association data account for ≈90% of passenger travel itineraries on commercial flights, excluding transportation via unscheduled charter flights; the remaining data are modeled using market intelligence.
Overview of the Cholera Outbreak in Malawi
Figure 1
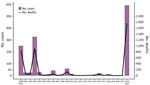
Figure 1. Cumulative case counts and deaths from cholera in Malawi by year, 1998–2023. Outbreaks occurred in 1998–1999, 2001–2002, 2008–2009, and 2022–2023. Scales for the y-axes differ substantially to underscore patterns but...
Before 2022, Malawi had experienced major cholera outbreaks in 1998–1999, 2001–2002, and 2008–2009 (5,7) (Figure 1). On March 3, 2022, Malawi declared a cholera outbreak after a confirmed case (symptom onset February 25, 2022) was found in Machinga District after Tropical Storm Ana (January 2022) and Cyclone Gombe (March 2022) caused severe flooding with resulting displacement of populations with low immunity to cholera and limited access to clean water and sanitation (7). Initially confined to flood-affected areas, the outbreak spread to central and northern regions by August 2022 (7). At the initiation of this study, 58,577 confirmed cases and 1,756 deaths were reported (Table; Figure 1). Given the consistent high case-fatality rate (>3%) and continued spread, the Malawi government declared a public health emergency on December 5, 2022. From December on, cases surged again affecting all regions, including Blantyre and Lilongwe, the country’s 2 largest cities (7). We obtained 70 V. cholerae isolates from stool samples collected during December 2022–March 2023. A total of 67 from the southern (n = 44), central (n = 18) and northern (n = 5) regions were processed into libraries (Table). From these 67 isolates, we generated 49 high-quality, near-complete V. cholerae genomes (Appendix 2 Figure 1).
Global Origin and Timing of 2022–2023 Cholera Outbreak
Figure 2

Figure 2. Phylogenetic history of cholera outbreaks within Malawi shown as part of a time-scaled maximum likelihood global phylogeny of 2,778 cholera genomes. Clade branches are colored by the previous 12 introduction...
Figure 3

Figure 3. Maximum clade credibility phylogeny depicting the clade containing genomes from the 2022–2023 outbreak in Malawi (n = 89, sampled December 10, 2022–February 2023) with a basal clade of genomes sequenced...
Figure 4
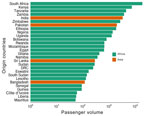
Figure 4. Number of passengers arriving by air travel into Malawi, by country of origin, January–June 2022. DRC, Democratic Republic of the Congo.
We constructed a time-scaled maximum-likelihood phylogeny based on high-quality SNPs of the 49 genomes from Malawi obtained in this study, 40 publicly available genomes (that passed quality filter) from the same outbreak (19), and 2,689 worldwide genomes (Figure 2). The genomes of the Malawi strains from this study clustered within a well-supported monophyletic clade (bootstrap >90%) together with the previously reported genomes from the same outbreak (19), denoting AFR15 introduction (20) (Figure 2). Those strains did not cluster with historical strains obtained from previous outbreaks within Malawi, which would indicate a new single introduction of cholera into the country. Instead, they were closely related to isolates from the 2022 outbreak in Pakistan (37) that clustered at the base of the clade, suggesting that the Malawi outbreak may have been caused by a strain introduction from Pakistan. However, we cannot reject that another unsampled country may have been involved in the origin of the Malawi cholera strain. The outbreak in Pakistan began in January 2022 and recorded >335,000 suspected cholera cases during January 15, 2022‒March 15, 2023 (38). Phylogeographic analyses confirmed movement of cholera from Pakistan into Malawi (BF = 180.7) (Appendix 2 Table 3). The maximum clade credibility tree estimated tMRCA of the Malawi strains to be July 4, 2022 and determined a 95% highest posterior density (HPD) interval of June 11–August 6 (Figure 3). The tMRCA shared between Pakistan and Malawi outbreaks was June 4, 2022 (95% HPD interval of March 24–July 1, 2022), which suggests that the Malawi outbreak may have resulted from a long-range transmission event from Pakistan as early as March 2022, a close estimate to the beginning of the outbreak (7). Because the spread of cholera in Africa is associated with human movement (9), we queried the passenger volumes originating from international airports and arriving in Malawi (Figure 4). We found that Malawi was well connected to Pakistan by large numbers of air passengers traveling between them during the first half of 2022 (Figure 4).
Cholera Transmission across Malawi and Africa
Figure 5
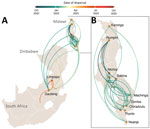
Figure 5. Spatiotemporal reconstruction of the spread of cholera in Malawi, South Africa, and Zimbabwe (A) and within Malawi (B) during the 2022–2023 outbreak. Circles represent nodes of the maximum likelihood phylogeny,...
Next, we explored the spread of cholera within Malawi. Our phylogeographic analysis supported a single source introduction into Malawi with consequent spread within the country and confirmed epidemiologic reports indicating that the outbreak expanded from southern regions, initially affected by flooding, to the northern and central parts of the country (Figure 5). The analysis showed evidence of dispersal from multiple hubs, including Machinga Blantyre, and Lilongwe, in the epidemic acceleration phase (Figure 5). Introduction of cholera from Malawi into South Africa occurred through multiple transmission events (Figure 5). Phylogeographic analyses strongly suggested that cholera spread into South Africa from Malawi (BF = 50,222) on multiple separate introductions (Appendix 2 Table 3). The substantial flow of air passengers between Malawi and South Africa underscores the strong connectivity between the countries (Figure 4). After the introduction in South Africa, cholera futher spread into Zimbabwe from South Africa (BF = 25,109) on >2 separate occasions (Figure 5). Our analysis was limited by the sampling date range (September 2022–September 2023) and thus might not provide accurate insights into the dispersal patterns during the early phase of the outbreak.
Effect of Severe Flooding in Malawi
Figure 6

Figure 6. Relationships between confirmed cholera cases and cholera outbreak dynamics in Malawi during the 2022–2023 outbreak. A) Cyclones and flooding events affecting Malawi are shown. Green dots represent flooding conditions across...
Because the 2022 Pakistan cholera outbreak was exacerbated by major floods during June‒October 2022 (39), we next examined the Malawi outbreak in light of the extreme weather events that occurred in 2022 (Figure 6, panel A; Appendix 2 Figure 2). The outbreak started after Tropical Storm Ana (January 28, 2022), which caused widespread flooding and damage in southern Malawi and left >200,000 persons displaced, many in emergency camps without adequate access to safe water and sanitation (7). Soon after the start of the outbreak, Cyclone Gombe caused further flooding (as measured by area flooded) and heavy damage across southern Malawi, including many areas already affected by Tropical Storm Ana (Appendix 2 Figure 2). The interval of the introduction of cholera into Malawi (95% HPD March 2022–August 2022) correlates with the beginning of the reported cases for the current cholera outbreak in the country (Figure 6, panel A; Appendix 2 Figure 2) and overlaps with flooding in Pakistan that exacerbated the ongoing cholera outbreak in that country. Initial floods in Malawi did not seem to correlate with amplification of cases, suggesting that the introduction was followed only by low circulation restricted to the flood-affected areas in the southern region (Appendix 2 Figure 2). Floods that occurred during the normal rainy season in late November 2022 likely intensified transmission; cases surged particularly in Blantyre and Lilongwe, the 2 main urban centers (Appendix 2 Figure 2) (7). The rapid surge of cases was likely spurred on by the extreme flooding events causing a lack of access to safe drinking water, poor sanitation and hygiene, and displacement of a vulnerable population (Figure 6) (7). Because we did not have genomes from the early phase of the outbreak in our study and because the introduction interval was relatively wide, spanning several months, we must consider an alternative hypothesis: the initial floods may have led to low-level circulation of endemic cholera strains, initiating the outbreak in Malawi. Subsequently, during the dry season, the introduction of the strain from Pakistan occurred with minimal transmission until later floods permitted the amplified transmission of a potentially more transmissible strain.
We estimated that Rt from daily case data increased after November 28, 2022, to a maximum of 1.619 on December 29, 2022, before starting to decline in early January 2023; it was consistently <1 from mid-February 2023 and at 0.704 at the end of the study period (Figure 6, panel B). Flooding (Figure 6, panel A) was positively associated with Rt (0.262 [95% CrI 0.003–0.509]) (Appendix 2 Figure 3, panel D).
Two oral cholera vaccination campaigns have been conducted since the onset of the outbreak; a total of 2,825,229 doses were administered by December 2, 2022, covering 96.8% of the population residing in communities with high risk and burden of cholera (7). We estimated a negative association between the vaccination campaigns and Rt (−0.320 [90% CrI −0.638 to −0.013]) (Appendix 2 Figure 3, panel D) but note that the association is only significant at a 90% CrI. Our results were robust to a varying level of the initial susceptible population (S0), which is important because the initial vaccination campaign in early 2022 may have reduced the S0 further than the S0 we used as our baseline, and we do not know about any prior immunity in the community caused by previous outbreaks (Appendix 2 Figure 3).
In this genomic analysis, we provide evidence of a link between the recent large cholera outbreaks in Pakistan and Malawi, consistent with previous genomic analyses revealing the importance of long-range V. cholerae transmission events between Asia and Africa (8,9). However, we cannot rule out that another unsampled country could be the origin of both the Malawi and Pakistan outbreaks. Our main findings support another recent analysis of the Malawi outbreak (19), which was linked to the strains previously identified in Asia, and another report of cases from South Africa in 2023 epidemiologically linked to Malawi (20). The extreme weather events and humanitarian crises in Malawi provided a suitable environment for the amplified spread of V. cholerae, and the subsequent movement of large numbers of persons may have enabled its spread to susceptible populations in areas relatively unaffected by cholera for more than a decade.
The relatively narrow sampling date range for this genomic analysis meant that we cannot confidently differentiate whether the introduction of V. cholerae was responsible for initiating the outbreak or if it only contributed to the later expansion of the outbreak through multiple transmission chains. Further sequencing of isolates from earlier stages of the outbreak could elucidate the initial dynamics, emphasizing the criticality of controlling cholera outbreaks and preventing introductions during the dry season to prevent escalation during subsequent flooding seasons. We are also working with partners in other heavily affected countries in Africa to conduct genomic sequencing of V. cholerae isolates, which will help with understanding the extent of cross-border regional transmission at different phases of the outbreak.
We modeled Rt over the course of the outbreak, incorporating satellite data to assess the effect of flooding on cholera transmission, which is particularly important in studying a waterborne disease like cholera. A substantial positive association between flooding and reproduction numbers explained the sustained high case numbers in January. The decline after February 2023 was likely caused by a successful vaccination campaign, infection-acquired immunity, and depletion of susceptible persons. Our model, based on national case data, lacked precise vaccination rollout details, which limited its accuracy. Future research should use spatially disaggregated data to better assess the effects of flooding and vaccination on cholera dynamics. Overall, our study highlights the need for coordinated global and regional cholera prevention and control efforts and the importance of heightened awareness, data sharing, and preparedness whenever outbreaks occur in any part of the world (40).
Dr. Chabuka is a dedicated medical scientist at the Public Health Institute of Malawi and a PhD fellow at Stellenbosch University. His research focuses on infectious diseases, including COVID-19, cholera, malaria, and arbovirus-related illnesses. His primary research interest is leveraging next-generation sequencing technologies to track pathogen evolution, antimicrobial resistance, and outbreak dynamics.
Acknowledgments
This article was preprinted at https://www.medrxiv.org/content/10.1101/2023.08.22.23294324v1.
With permission from the Public Health Institute of Malawi, raw reads of the isolates that were successfully sequenced have been made publicly available and deposited in the National Center for Biotechnology Information Short Read Archive (BioProject PRJNA967700).
Centre for Epidemic Response and Innovation and the CLIMADE program are supported by grants from the Rockefeller Foundation (no. HTH 017), the SAMRC South African mRNA Vaccine Consortium, the South African Department of Science and Innovation, and by Medical Research Foundation (no. MRF-RG-ICCH-2022-100069). S.B. and C.M. acknowledge funding from the MRC Centre for Global Infectious Disease Analysis (reference no. MR/X020258/1), funded by the UK Medical Research Council. This UK-funded award is carried out in the frame of the Global Health EDCTP3 Joint Undertaking. S.B. is funded by the National Institute for Health and Care Research (NIHR) Health Protection Research Unit in Modelling and Health Economics, a partnership between UK Health Security Agency, Imperial College London, and London School of Hygiene & Tropical Medicine (grant code NIHR200908); the Novo Nordisk Foundation via the Novo Nordisk Young Investigator Award (no. NNF20OC0059309); and the Danish National Research Foundation (no. DNRF160). S.B. acknowledges support from the Eric and Wendy Schmidt Fund For Strategic Innovation via the Schmidt Polymath Award (no. G-22-63345) , which also supports C.M.
The views expressed are those of the authors and not necessarily those of the NIHR, UK Health Security Agency or the UK Department of Health and Social Care. The funders of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report.
References
- Kanungo S, Azman AS, Ramamurthy T, Deen J, Dutta S. Cholera. Lancet. 2022;399:1429–40. DOIPubMedGoogle Scholar
- World Health Organization. Outbreaks and other emergencies bulletin, week 30: 24–30 July 2023. 2023 [cited 2025 Feb 24]. https://www.afro.who.int/health-topics/disease-outbreaks/outbreaks-and-other-emergencies-updates
- World Health Organization. Disease outbreak news; cholera—global situation. 2022 Dec 16 [cited 2025 Feb 24]. https://www.who.int/emergencies/disease-outbreak-news/item/2022-DON426
- World Health Organization. Disease outbreak news; cholera—global situation. 2023 Feb 11. [cited 2025 Feb 24]. https://www.who.int/emergencies/disease-outbreak-news/item/2023-DON437
- Msyamboza KP, Kagoli M, M’bang’ombe M, Chipeta S, Masuku HD. Cholera outbreaks in Malawi in 1998‒2012: social and cultural challenges in prevention and control. J Infect Dev Ctries. 2014;8:720–6. DOIPubMedGoogle Scholar
- World Health Organization African Region. Weekly regional cholera bulletin. 2024 Apr 1 [cited 2025 Feb 24]. https://iris.who.int/bitstream/handle/10665/376526/AFRO%20Cholera%20Bulletin.58.pdf
- World Health Organization. Disease outbreak news: cholera—Malawi. 2023 Feb 9 [cited 2025 Feb 24]. https://www.who.int/emergencies/disease-outbreak-news/item/2022-DON435
- Mutreja A, Kim DW, Thomson NR, Connor TR, Lee JH, Kariuki S, et al. Evidence for several waves of global transmission in the seventh cholera pandemic. Nature. 2011;477:462–5. DOIPubMedGoogle Scholar
- Weill FX, Domman D, Njamkepo E, Tarr C, Rauzier J, Fawal N, et al. Genomic history of the seventh pandemic of cholera in Africa. Science. 2017;358:785–9. DOIPubMedGoogle Scholar
- Inzaule SC, Tessema SK, Kebede Y, Ogwell Ouma AE, Nkengasong JN. Genomic-informed pathogen surveillance in Africa: opportunities and challenges. Lancet Infect Dis. 2021;21:e281–9. DOIPubMedGoogle Scholar
- Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–90. DOIPubMedGoogle Scholar
- Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–93. DOIPubMedGoogle Scholar
- Tonkin-Hill G, Lees JA, Bentley SD, Frost SDW, Corander J. Fast hierarchical Bayesian analysis of population structure. Nucleic Acids Res. 2019;47:5539–49. DOIPubMedGoogle Scholar
- Croucher NJ, Page AJ, Connor TR, Delaney AJ, Keane JA, Bentley SD, et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2015;43:
e15 . DOIPubMedGoogle Scholar - Shen W, Le S, Li Y, Hu F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One. 2016;11:
e0163962 . DOIPubMedGoogle Scholar - Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol Biol Evol. 2018;35:1547–9. DOIPubMedGoogle Scholar
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74. DOIPubMedGoogle Scholar
- Sagulenko P, Puller V, Neher RA. TreeTime: maximum-likelihood phylodynamic analysis. Virus Evol. 2018;4:
vex042 . DOIPubMedGoogle Scholar - Chaguza C, Chibwe I, Chaima D, Musicha P, Ndeketa L, Kasambara W, et al. Genomic insights into the 2022–2023 Vibrio cholerae outbreak in Malawi. Nat Commun. 2024;15:6291.
- Smith AM, Sekwadi P, Erasmus LK, Lee CC, Stroika SG, Ndzabandzaba S, et al. Imported cholera cases, South Africa, 2023. Emerg Infect Dis. 2023;29:1687–90. DOIPubMedGoogle Scholar
- Olatunji G, Kokori E, Moradeyo A, Olatunji D, Ajibola F, Otolorin O, et al. A perspective on the 2023 cholera outbreaks in Zimbabwe: implications, response strategies, and policy recommendations. J Epidemiol Glob Health. 2024;14:243–8. DOIPubMedGoogle Scholar
- Rambaut A, Lam TT, Max Carvalho L, Pybus OG. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016;2:
vew007 . DOIPubMedGoogle Scholar - Gill MS, Lemey P, Faria NR, Rambaut A, Shapiro B, Suchard MA. Improving Bayesian population dynamics inference: a coalescent-based model for multiple loci. Mol Biol Evol. 2013;30:713–24. DOIPubMedGoogle Scholar
- Suchard MA, Lemey P, Baele G, Ayres DL, Drummond AJ, Rambaut A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018;4:
vey016 . DOIPubMedGoogle Scholar - Baele G, Li WL, Drummond AJ, Suchard MA, Lemey P. Accurate model selection of relaxed molecular clocks in bayesian phylogenetics. Mol Biol Evol. 2013;30:239–43. DOIPubMedGoogle Scholar
- Mavian C, Paisie TK, Alam MT, Browne C, Beau De Rochars VM, Nembrini S, et al. Toxigenic Vibrio cholerae evolution and establishment of reservoirsin aquatic ecosystems. Proceedings of the National Academy of Sciences. 2020:201918763.
- Mavian CN, Tagliamonte MS, Alam MT, Sakib SN, Cash MN, Moir M, et al. Ancestral origin and dissemination dynamic of reemerging toxigenic Vibrio cholerae, Haiti. Emerg Infect Dis. 2023;29:2072–82. DOIPubMedGoogle Scholar
- Yu GC, Smith DK, Zhu HC, Guan Y, Lam TTY. GGTREE: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol. 2017;8:28–36. DOIGoogle Scholar
- Bielejec F, Rambaut A, Suchard MA, Lemey P. SPREAD: spatial phylogenetic reconstruction of evolutionary dynamics. Bioinformatics. 2011;27:2910–2. DOIPubMedGoogle Scholar
- Bhatt S, Ferguson N, Flaxman S, Gandy A, Mishra S, Scott JA. Semi-mechanistic Bayesian modelling of COVID-19 with renewal processes. J R Stat Soc Ser A Stat Soc. 2023;186:601–15. DOIGoogle Scholar
- Cori A, Ferguson NM, Fraser C, Cauchemez S. A new framework and software to estimate time-varying reproduction numbers during epidemics. Am J Epidemiol. 2013;178:1505–12. DOIPubMedGoogle Scholar
- Cartalis C, Feidas H, Glezakou M, Proedrou M, Chrysoulakis N. Use of earth observation in support of environmental impact assessments: prospects and trends. Environ Sci Policy. 2000;3:287–94.
- Tarpanelli A, Mondini AC, Camici S. Effectiveness of Sentinel-1 and Sentinel-2 for flood detection assessment in Europe. Nat Hazards Earth Syst Sci. 2022;22:2473–89. DOIGoogle Scholar
- Li S, Sun D, Goldberg MD, Sjoberg B, Santek D, Hoffman JP, et al. Automatic near real-time flood detection using Suomi-NPP/VIIRS data. Remote Sens Environ. 2018;204:672–89.
- Turner B. International Air Transport Association (IATA). In: Turner B, editor. The statesman’s yearbook 2008: the politics, cultures and economies of the world. London: Palgrave Macmillan UK; 2007. p. 48.
- Sim EM, Martinez E, Blackwell GA, Pham D, Millan G, Graham RMA, et al. Genomes of Vibrio cholerae O1 serotype Ogawa associated with current cholera activity in Pakistan. Microbiol Resour Announc. 2023;12:
e0088722 . DOIPubMedGoogle Scholar - World Health Organization Eastern Mediterranean Region. Epidemic and pandemic-prone diseases: acute watery diarrhoea/cholera updates (16–31 March 2023). 2023 [cited 2025 Feb 24]. https://www.emro.who.int/pandemic-epidemic-diseases/cholera/acute-watery-diarrhoeacholera-updates-1631-march-2023.html
- Jamil H, Liaqat A, Lareeb I, Tariq W, Jaykumar V, Kumar L, et al. Monsoon and cholera outbreaks in Pakistan: a public health concern during a climate catastrophe. IJS Global Health. 2023;6:e105.
- Global Task Force on Cholera Control. Ending cholera: a global roadmap to 2030. 2017 [cited https://www.gtfcc.org/wp-content/uploads/2019/10/gtfcc-ending-cholera-a-global-roadmap-to-2030.pdf
Figures
Table
Cite This ArticleOriginal Publication Date: May 20, 2025
1These authors contributed equally to this article.
Table of Contents – Volume 31, Number 6—June 2025
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Carla N. Mavian, University of Florida—Emerging Pathogens Institute, 2055 Mowry Rd , Gainesville, FL 32610 USA: Tulio de Olivera, JC Smuts building, School for Data Science and Computational Thinking, Stellenbosch University, Stellenbosch, South Africa
Top